1.Несколько файлов в формате fasta собрать в единый файл
Исходные файлы: здесь
Команда: seqret @mylist mysequences.fasta
Результат: здесь
2.Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы
Исходный файл: здесь
Команда: seqretsplit mysequences.fasta -auto
Результат: здесь
3.Из файла с хромосомой в формате .gb вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле
Исходные файлы: здесь
Команда: seqret @list three.fasta
Результат: здесь
4.Транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода. Результат - в одном fasta файле. 5.Транслировать данную нуклеотидную последовательность в шести рамках.
Исходный файл: здесь
Команда : transeq -sequence task4.fasta protein.fasta -frame 6 -table 11
Результат: здесь
6.Перевести выравнивание и из fasta формате в формат .msf
Исходный файл: здесь
Команда: seqret task6.fasta msf::task6.fasta
Результат: здесь
7. Выдать в выходной поток число совпадающих букв между второй последовательностью выравнивания и всеми остальными (на выходе только имя последовательности и число)
Исходный файл: здесь
Команда: infoalign task6.msf -outfile stdout -refseq 2 -only -name -idcount >task6.txt
Результат: здесь
8.Перевести аннотации особенностей в записи формата .gb в табличный формат .gff
Исходный файл: здесь
Команда: featcopy sequence.gb -outfeat task8.gff
Результат: здесь
9.Из данного файла с хромосомой в формате .gb получить fasta файл с кодирующими последовательностями; (*) добавить в описание каждой последовательности функцию белка (из поля product)
Исходный файл : здесь
Команда: extractfeat sequence.gb -describe product -type CDS -outseq task9.txt
Результат: здесь
10.Перемешать буквы в данной нуклеотидной последовательности.
Исходный файл: здесь
Команда: shuffleseq HS.fasta shuffled.fasta
Результат: здесь
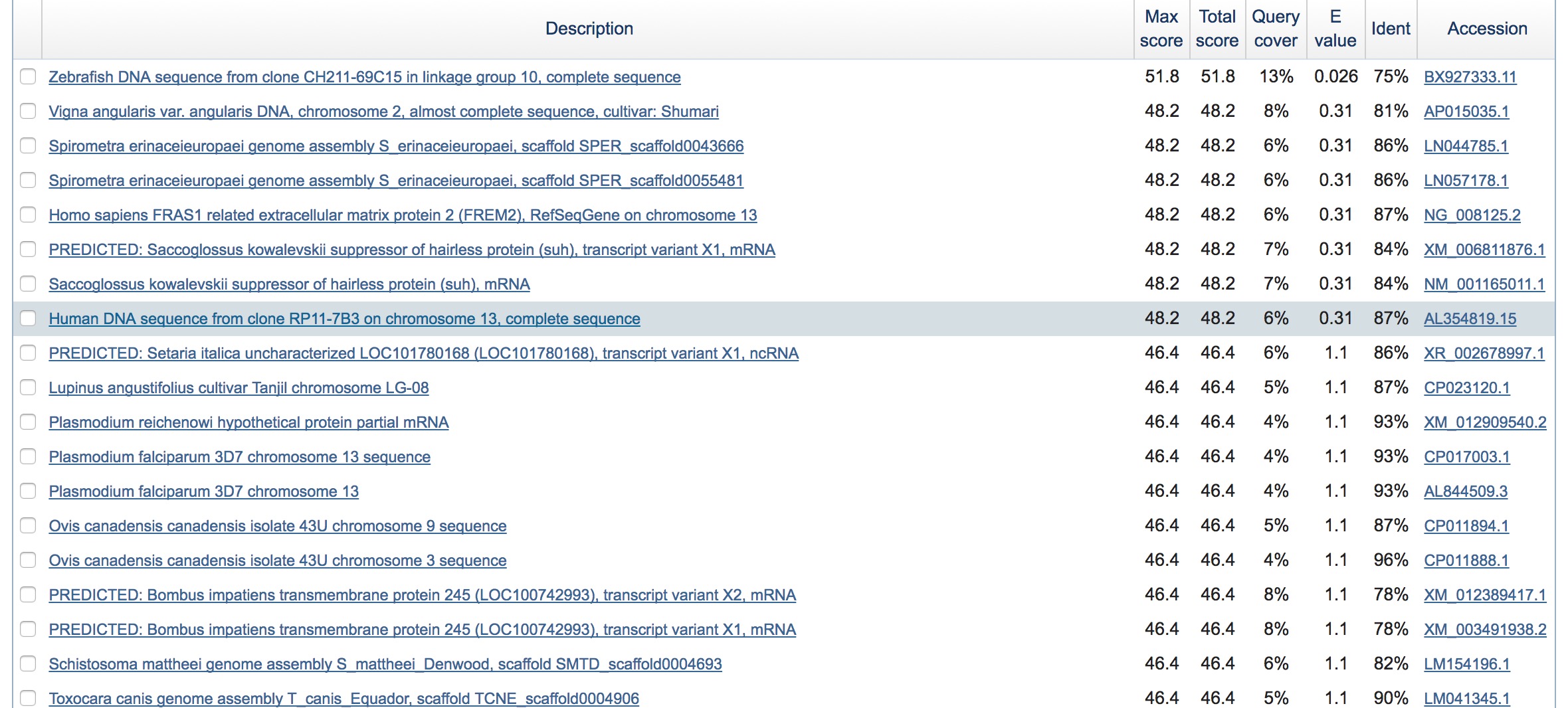
11.Для случайной последовательности проверить с помощью blastn сколько "достоверных" находок (с E-value < 0.1) найдется в нуклеотидном банке данных (запустите blastn с порогом E = 10 - по умолчанию и посчитайте сколько с E-value < 0.1)
Исходный файл: здесь
Параметры запуска blastn: E-value=10; wordsize:7; db: nt.
Результат:
12.Найдите все открытые рамки длиной более 200 в бактериальной хромосоме и посчитайте статистику совпадений с аннотированными кодирующими последовательностями белков.
Исходный файл: здесь и здесь
Команда: getorf -sequence sequence.fasta -outseq task12.txt -circular -table 11 -minsize 200 и featcopy chromosome.gb featcopy.txt
Результат: здесь и здесь. Число 200 было выбрано как одно из самых минимальных значений длин CDS в файле genebank. Данные были обработаны с помощью Excel. Программа getorf выдает отсортированные по порядку открытые рамки считывания, сначала на прямой, потом на комплементарной цепях. Причем по сравнению с данными, полученными из genebank они смещены на 3 нуклеотида. То есть к номеру стоп кодона из файла, поулченного geneorf нужно прибавить 3 для смысловой и вычесть 3 для комплементарной цепочки, чтобы сравнивать их со стоп кодонами, полученными из записи genebank. Таким образом программы geneorf нашла 23394 рамки считывания, а featcopy - 9578. Пересечения этих множеств (если учитывать только стоп кодоны, так как с определением инициаторного могут быть проблемы даже в записи) составляют 2194, или 22,91% от предсказанных в genebank. Ссылка на excel файл.
13.Найдите частоты кодонов в данных кодирующих последовательностях
Исходный файл: здесь
Команда: cusp ../task4/task4.fasta task13
Результат: здесь
14.Найдите частоты динуклеотидов в хромосоме человека, сравните их с ожидаемыми
Исходный файл: здесь
Команда: wordcount hs_ref_GRCh38.p7_chr22.fa -wordsize 1 -outfile task14.txt и wordcount hs_ref_GRCh38.p7_chr22.fa -wordsize 2 -outfile task14_2.txt
Результаты: здесь . Результаты программы wordcount были обработаны в Excel. Величина отклонения от ожидаемых значений больше всего у CG, TA, TG, CA.
15.Выровняйте кодирующие последовательности соответственно выравниванию белков - их продуктов
Файлы: здесь и здесь
Команда: tranalign -aseq genes.fasta -bseq proteinalign.fasta -out out.fasta
Результат: здесь
16.Постройте локальное множественное выравнивание трех нуклеотидных последовательностей
Исходные файлы: здесь
Команда:edialign @list -revcomp -outfile task16.fasta -outseq out.fasta
Результат: здесь и здесь
17.Удалите символы гэпов и другие посторонние символы из последовательности.
Исходные файлы: здесь
Команда:degapseq out.fasta task17.fasta
Результат: здесь
18. Переведите символы конца строки в формат unix
Исходные файлы: здесь
Команда: noreturn 1.txt task18
Результат: здесь
19.Создайте три случайных нуклеотидных последовательностей длины сто
Команда: makenucseq -amount 3 -length 100 -outseq task19.fasta
Результат: здесь
20.Файл с ридами sra_data.fastq в формате fastq перевести в формат fasta.
Исходные файлы: здесь
Команда: seqret sra_data.fastq fasta::alignment.fasta
Результат: здесь