Поиск сигналов
Задание 1. Сравнение состава систем рестрикции модификации, закодированных в двух штаммах вида Klebsiella oxytoca
В данном задании было необходимо сравнить состав систем рестрикции-модификации у двух представителей вида Klebsiella oxytoca: у одного использовался полный геном из NCBI (идентификатор: CP008788.1), а у другого - набор контигов из метагенома кишечника человека.
Большинство известных сайтов рестрикции-модификации представлены в файле.
Для определения того, какие из имеющихся сайтов присутствуют у интересующей бактерии, производился подсчет контраста: отношение наблюдаемой частоты встречаемости сайта к ожидаемой. Так как известно, что, несмотря на то, что сайты рестрикции в бактериальном геноме метилированы и не должны разрезаться, иногда случаются ошибки, поэтому сайты систем рестрикции-модификации должны встречаться в бактериальном геноме реже ожидаемого.
В данном случае подсчет контраста производился по методу Карлина, а пороговое значение контраста было определено в 0.78 (т. е. значение контраста такое и меньше свидетельствует о присутствии данной системы РМ у бактерии).
Подсчет контраста выполнялся с помощью веб-вервиса.
Выходной файл для полного генома бактерии: full.
Выходной файл для бактерии из метагенома кишечника: meta.
Файл с отобранными избегаемыми сайтами: clear.tsv.
Для удобства в Таблице 1 перечислены избегаемые сайты для обоих представителей.
Таблица 1. Избегаемые сайты (расчет для полного генома бактерии Klebsiella oxytoca и для набора контигов бактерии данного вида из организма человека).
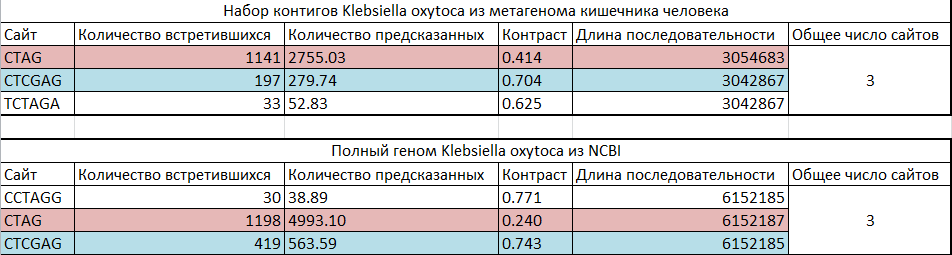
Как видно из Таблицы 1, оба штамма имеют по три системы РМ, причем 2 из них совпадают (выделены розовым и голубым цветами).
Во-первых, думаю, что на результат каким-то образом повлиял тот факт, что в одном случае у нас полный геном, а в другом - набор контигов. Видно, что в случае набора контигов последовательность в два раза короче. И если для сайта CTCGAG реальная и ожидаемая встречаемости пропорциональны длине, т. е. контраст приблизительно одинаковый, то для сайта CTAG контраст в случае метагенома в 2 раза больше.
Что можно сказать по поводу таких результатов? Изначально я предполагала, что у бактерии с полным геномом будет больше систем РМ, так как бактерия в кишечнике лучше защищена, поэтому ей не нужно так много систем. Но результаты не подтвердили эту теорию, а найти данные об условиях существования бактерии с полным геномом не удалось.
Задание 2. Поиск последовательности Шайн-Дальгарно в геноме Aquifex aeolicus
Бесконечный поток разочарований на этом не прервался.
В этом задании требовалось найти в геноме своей бактерии (Aquifex aeolicus VF5) последовательности Шайна-Дальгарно - сайты инициации трансляции у прокариот.
По данным статей, эта последовательность в общем случае составляет 5-6 нуклеотидов и расположена приблизительно в позициях -10 - -5 относительно старта трансляции [1] (в другой статье было указано, что старт SD обычно расположен в позиции -7 относительно старта инициации [2]). В статье о полном геноме данной бактерии ШД не упоминается [3]. Однако мне удалось найти интересные сведения о ШД в геноме родственной бактерии Thermophillus thermophillus (в предыдущих заданиях мне часто приходилось пользоваться этим организмом как источником гомологичных структур и свойств). На Рис. 1 изображена гистограмма расстояний от старта трансляции до начала SD в этом организме [4].
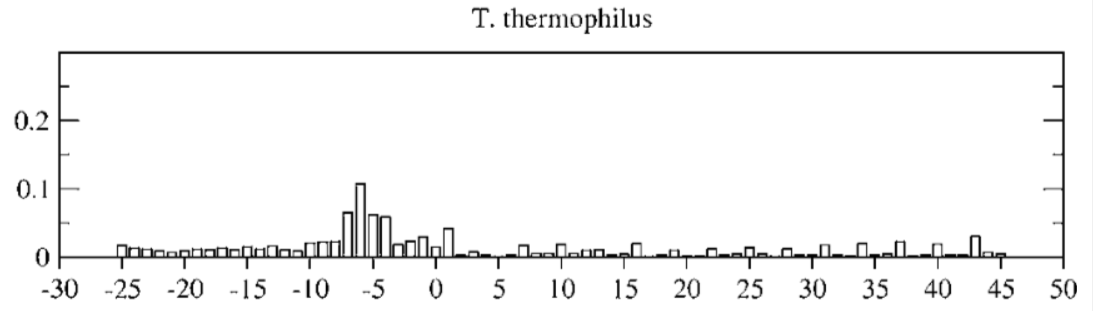
На основе этих данных я предложила для поиска SD участки последовательностей в позициях -15 - -1 относительно стартов трансляции (AUG) генов моей бактерии. Для этого было выбрано около 550 хороших генов (длина больше 300 п.н., аннотированные функции). Далее я производила запуск сервиса МЕМЕ, где указала дополнительные параметры поиска: ограничила поиск 1 мотивом, длину мотива - 6 нуклеотидами, мотив искался только на данной цепи, было достаточно найти не менее 150 мотивов (SD встречается около не у всех генов, регуляция не так проста). Главным результатом работы сервиса является позиционно-весовая матрица, которая будет использоваться для поиска мотива в другом месте.
PWM:
Motif 1 position-specific probability matrix
--------------------------------------------------------------------------------
letter-probability matrix: alength= 4 w= 7 nsites= 168 E= 7.7e-165
0.559524 0.154762 0.172619 0.113095
0.083333 0.029762 0.833333 0.053571
0.000000 0.000000 1.000000 0.000000
0.988095 0.000000 0.011905 0.000000
0.000000 0.000000 1.000000 0.000000
0.000000 0.000000 1.000000 0.000000
0.178571 0.041667 0.101190 0.678571
Затем уже проводился поиск по регионам перед всеми генами с помощью ресурса FIMO. В этом случае были взяты более широкие участки: от -25 до +6 (если посмотреть на Рис. 1, там довольно высокий пик в области +1 нуклеотида - кстати, статья в целом посвящена необычной инициации трансляции, поэтому как раз изобилует примерами SD внутри области гена). Также я пробовала запускать ресурс с разными параметрами E-value: 0,1 и 0,01. При значении 0.1 выдача слишком большая, и ясно, что многие находки не относятся к SD.
Выходной файл FIMO в формате Excel (Лист 1) с добавленным расстоянием до старта трансляции (подсчитано на Листе 2): Excel.
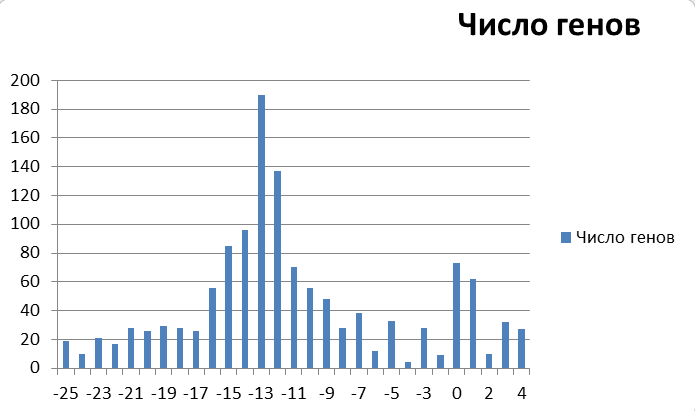
На Рис. 2 - гистограмма расстояний от старта трансляции до начала SD.
На Рис. 3 приведен лого данного мотива.
Процент генов, перед которыми была найдена SD: около 80%.
Процент генов получился даже больше, чем бывает в реальности. Возможно, это связано с шириной участка, выбранного для поиска, однако, лого очень хороший, поэтому есть вероятность, что результатам можно доверять. Я также читала информацию о том, что не считая расположения SD внутри транслируемой области, для моей бактерии, в целом, характерны типичные расположение и консенсус [5], что и подтверждается результатами данного практикума.
Использованные источники:
[1] Wikipedia
[2] Determination of the optimal aligned spacing between the Shine-Dalgarno sequence and the translation initiation codon of Escherichia coli mRNAs
[3] The complete genome of the hyperthermophilic bacterium Aquifex aeolicus
[4] Predicting Shine–Dalgarno Sequence Locations Exposes Genome Annotation Errors
[5] An Exact Method for Finding Short Motifs in Sequences, with Application to the Ribosome Binding Site Problem