| Главная страница | Семестры | О себе | Ссылки |


В данном практикуме я работала с седьмой хромосомой человека (сборка hg19). Ссылку на саму хромосому давать не буду (боюсь - очень большой объем), но вот одноконцевые чтения.
Анализ качества чтений
Контроль качества чтений был осуществлен при помощи программы FastQC, ее вызов на рисунке 1. Результат работы на рисунке 2.
Рис. 1. Вызов FastQC
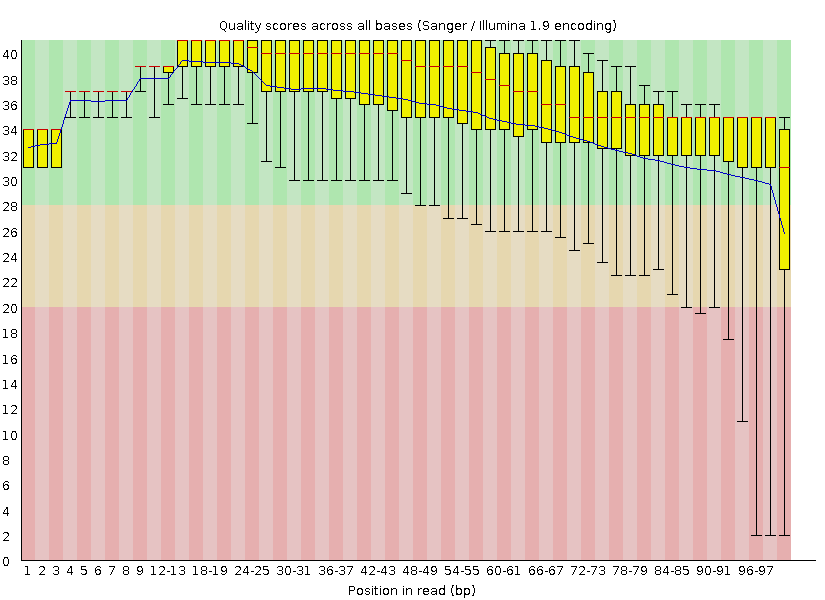
Рис. 2. Выдача FastQC. Видно, что большинство ридов (особенно в начале) в зеленой области, то есть качество прочтения хорошее, но не для всех ридов
Очистка чтений
Очистка была проведена программой Trimmomatic: с конца каждого чтения были отрезаны нуклеотиды скачеством ниже 20, удалены риды короче 50. Команда представлена на рисунке 3.
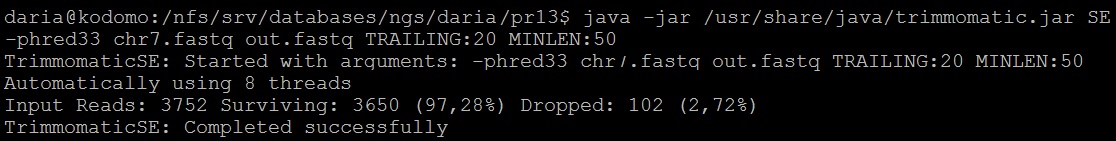
Рис. 3. Очистка чтений
Было удалено 102 рида, осталось 3650 ридов (97,28% от исходного количества).
После этого был снова сделан контроль качества чтений с помощью FastQC (на рисунке 4 команда, на рисунке 5 - результат).
Рис. 4. Повторный вызов FastQC
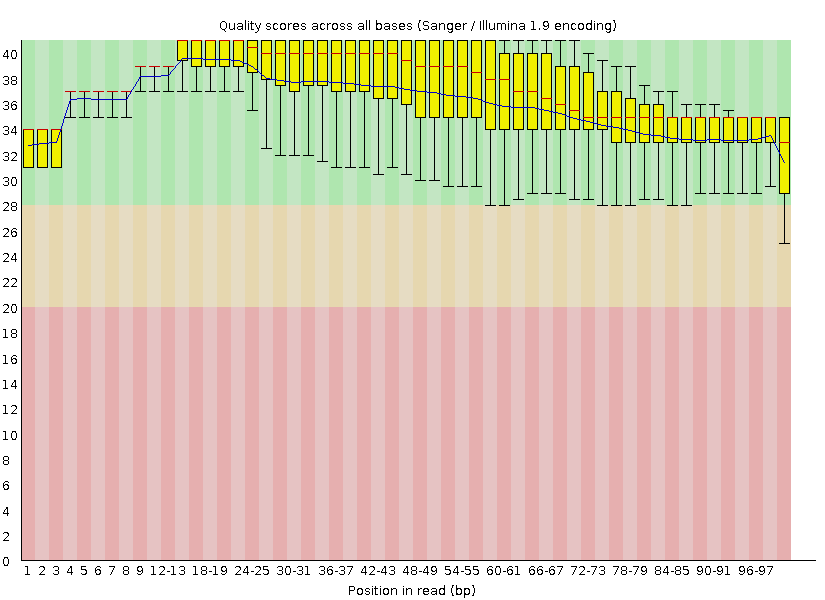
Рис. 5. Вторая выдача FastQC. Все риды качественно прочтены
Картирование чтений
С помощью программы BWA очищенные чтения были откартированы. То есть сначала была проиндексирована референсная последовательность - командой с рисунка 6.
Рис. 6. Индексирование референсной последовательности
Затем было построено выравнивание прочтений и референса в формате .sam - на рисунке 7.
Рис. 7. Построение выравнивания прочтений и референса
Анализ выравнивания
Требовалось перевести полученное выравнивание в бинарный формат .bam, для этого использовался пакет samtools, как показано на рисунке 8.

Рис. 8. Форматирование выравнивания
Затем с помощью приведенной на рисунке 9 команды содержание переформатированного файла было отсортировано по координате в рефересе начала чтения, далее проиндексировано (рисунок 10). На рисунке 11 - выяснение того, сколько ридов в итоге откартировалось на геном.

Рис. 9. Сортировка содержимого файла

Рис. 10. Индексирование содержимого файла

Рис. 11. Подсчет откартированных ридов
Откартировалось 3648 ридов, что меньше исходного количества ридов после очистки. Я не знаю механизма работы использованных мной программ, поэтому не могу сказать ни почему так получилось, ни на каком этапе.
Поиск SNP и инделей
И вот наконец можно перейти к собственно поиску однонуклеотидных полиморфизмов (SNP) - отличий в один нуклеотид между одинаковыми местами геномов представителей одного вида или между гомологичными участками гомологичных хромосом.
Был создан файл с полиморфизмами в формате .bcf, как показано на рисунке 12, а затем файл с отличиями между референсом и чтениями в формате .vcf, как на рисунке 13.

Рис. 12. Создание файла с полиморфизмами
Рис. 13. Создание файла со списком отличий
В таблице 1 приведены примеры полиморфизмов из файла .vcf.
Таблица 1. Однонуклеотидные полиморфизмы
Координата |
Полиморфизм |
Референс (REF) |
Чтения (ALT) |
Глубина чтений (DP) |
Качество чтений (QUAL) |
134237294 |
Замена |
А |
G |
1 |
6,20226 |
134261097 |
Замена |
C |
G |
66 |
225,009 |
5122854 |
Вставка |
ATT |
ATTT |
8 |
105,467 |
Анализ SNP
Из файла с отличиями были удалены все индели, c помощью предоставленного скрипта фомат был изменен на пригодный для дальнейшей работы (рисунок 14). Проведено аннотирование по различным базам данных (рисунки 15-19). Полученная информация собрана в итоговой таблице.

Рис. 14. Переформатирование файла с отличиями
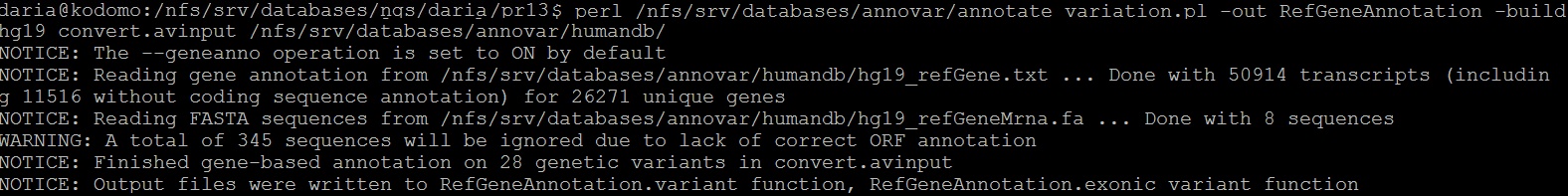
Рис. 15. Аннотация по RefGene

Рис. 16. Аннотация по snp138

Рис. 17. Аннотация по 1000 genomes

Рис. 18. Аннотация по Gwas
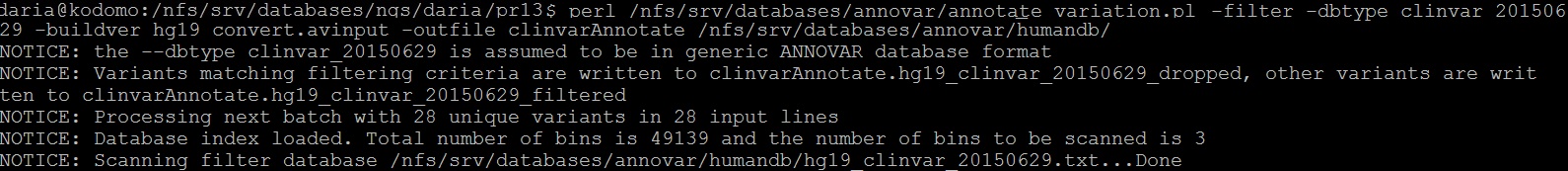
Рис. 19. Аннотация по Clinvar
Выводы:
Было найдено 28 полиморфизмов (из них аннотировано 25), 3 инделя.
25 SNP имеют свои rs (по базе snp138).
Полиморфизмы привели к различным нуклеотидным заменам, закономерности в которых я не вижу. К примеру, один из гуанино в гене ACHE был заменен на аденин (G на позиции 100490077), а в гене WNT16 два цитозина были заменены на тимины (позиции 120965652 и 120979089).
База данных RefSeq делит полиморфизмы на несколько категорий: exonic, splicing, ncRNA, UTR5, UTR3, intronic, upstream, downstream, intergenic. В моем случае к exonic относятся 4 SNP, к UTR3 - 2, intronic - 22, предствителей иных категорий найдено не было.
Для 25 полиморфизмов (всех) известна частота втречаемости аллелей (по базе 1000 genomes).
4 полиморфизма имеют клиническое значение: влияют на развитие диабета 2-го типа, содержание минералов в костной ткани, толщину кортикального слоя и продолжительность жизни.
Не было найдено SNP, имеющих влияние на фенотип (по базе Сlinvar).
14 snp имеют покрытие больше 10, что считается хорошим покрытием, и имеют высокое качество чтений. 7 snp имеют покрытие от 5 до 10 (включительно), что считается неплохим покрытием, и имеют хорошее качество чтений.7 snp имеют покрытие меньше 5, что считается плохим покрытием и имеют низкое качество чтений. Из них 5 SNP имеют покрытие равное 1, что считается очень плохим покрытием и имеют качество чтений меньше 7 (или близкое к 7, как в случае snp с координатой 134255326), что заставляет задуматься, действительно ли это полиморфизмы.
Полиморфизмы попали на известные гены: ACHE, WNT16, AKR1B15 (по базе Gwas).
18 SNP достаточно распространены (частоты встречаемости от 0,7 до 3), 7 редкие (частоты встречаемости меньше 0,1).
© Дарья Горбачева | изменено 16.03.2016 |