Практикум 12
1. Сравнение выравниваний разными программами MSA
В качестве референсного выравнивания я выбрал выравнивание из базы данных идеальных выравниваний BAliBASE - B30001.
С идеальным выравниванием сравнивались 3 программы множественного выравнивания: ClustalO; Mafft; Muscle, чтобы сравнить результаты был использована программа, написанна я с помощью Python, результаты ее работы представлены ниже.Ссылка на проект Jalview ; Совпавшие колонки для каждой из программ: Muscle; Mafft; Clustal
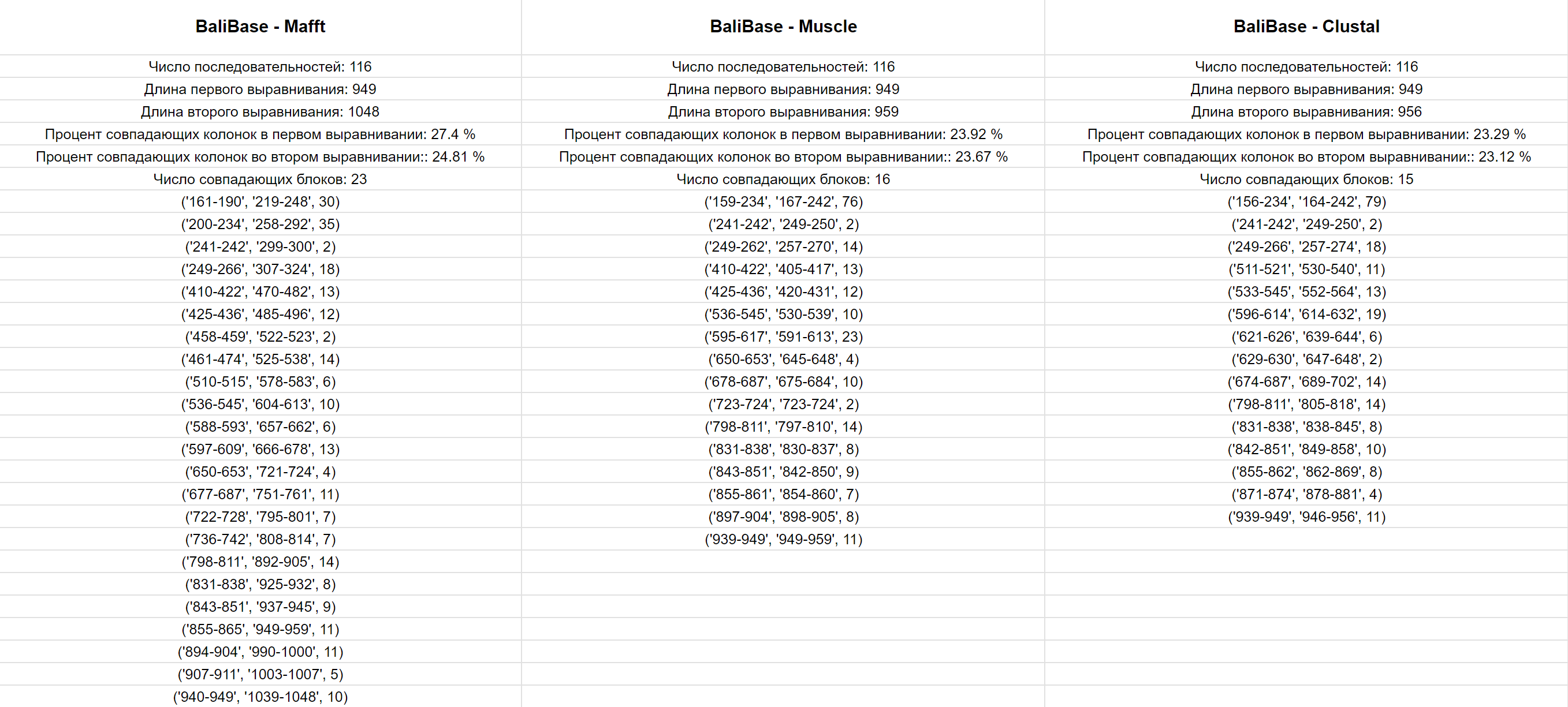
Из таблицы видно, что во всех трех случаях процент совпадающих колонок довольно низкий: максимальный процент - 27,4% (Mafft). При этом самый длинный совпадающий блок был обнаружен программой Clustal - 79 позиций, также несмотря на то, что есть выравнивание и с большим процентом совпадения колонок, выравнивание Clustal по своей длине самое самое близкое к идеальному (референсному). Таким образом Mafft выдал больше совпадающих блоков, но они относительно короткие и сама длина выравнивания больше на 100 позиций, чем референсное, что составляет около 10% от все последовательности, это может сказываться на достоверности выравнивания, а Muscle и Clustal сработали практически одинаково, но в силу того, что длина выравнивания Clustal немного ближе к референсному, самым он и является самой точной программой MSA.
2. 3D выравнивание
Для 3D выравнивания был выбран домен из PFam: PF00265 Thymidine kinase
Пространственное выравнивание осуществлялось с помощью Pairwise Structure Alignment на сайте PDB алгоритмом TM-align, обычное выравнивание производилось с помощью программы Mafft. Референсной была структура 2JA1 - Thymidine kinase from B. cereus with TTP bound as phosphate donor, две другие структуры - 3E2I - Crystal structure of Thymidine Kinase from S. aureus; 2J9R - Thymidine kinase from B. anthracis in complex with dT
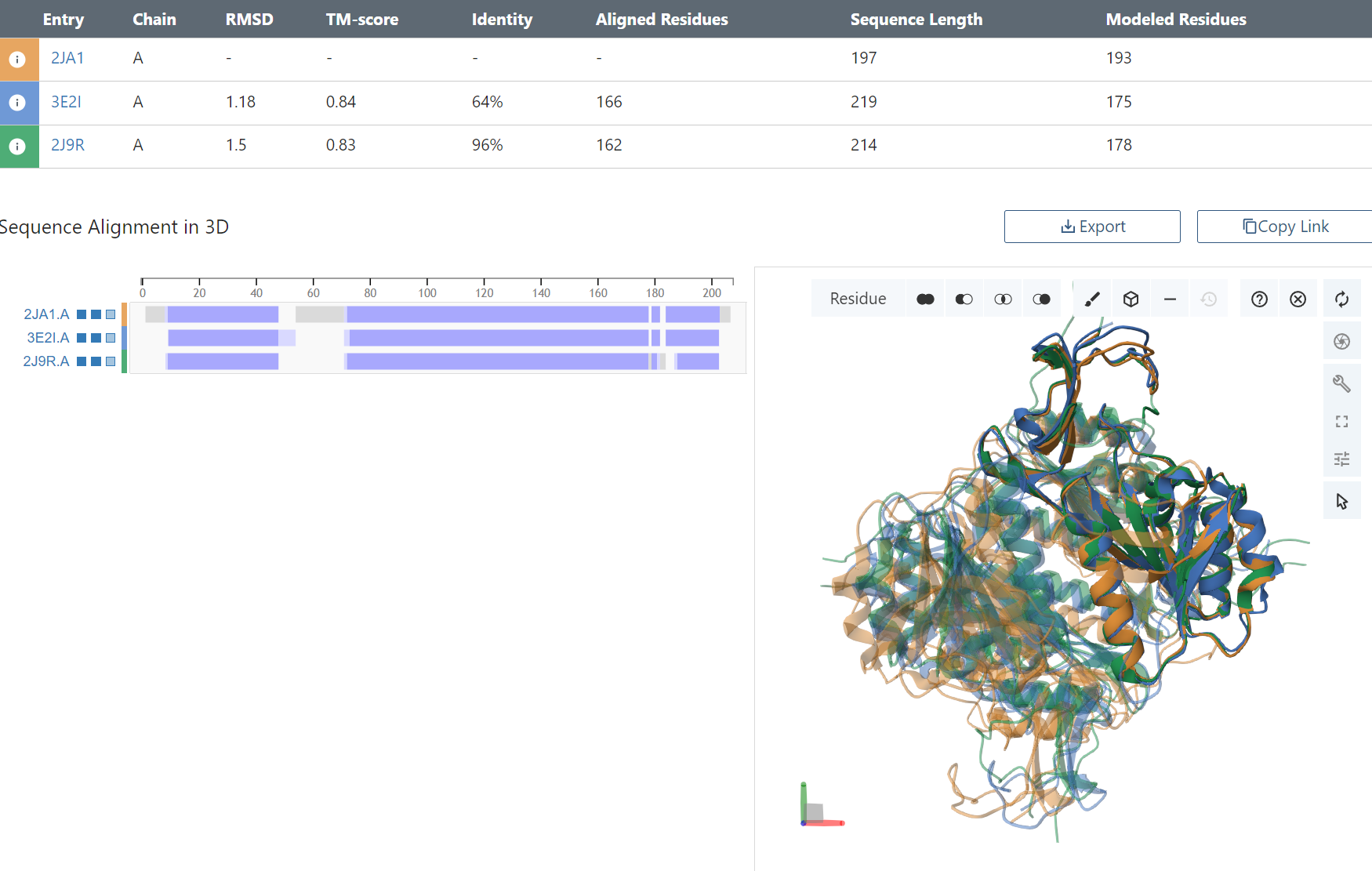
Из выравниваний (3D и Clustal) можно сделать вывод о том, что три выбранные структуры имеют много идентичных участков, из чего можно предположить, что данные киназы схожи между собой, довольно много консервативных участков: (10-13);(17-30);(89-96);(112-119);(140-147);(155-161);(169-177);(179-186) - это лишь часть из совпадающих блоков. Если говорить про результаты 3D выравнивания, то можно сказать, что у пары 2JA1 и 2J9R больше консервативных участков, чем у пары 2JA1 и 3E2I, также у этих пар гэпы встречаются не всегда в одних и тех же местах, возможно это связано с различием в геометрии молекулы между вторичными структурами (в основном спиралями).
3. Описание работы программы Mafft
MAFFT это программа, которая используется для создания множественных выравниваний нуклеотидных и аминокислотных последовательностей. Изначально использовался алгоритм, основанный на прогрессивном выравнивании при помощи быстрого преобразования Фурье. В более поздних версиях программы были добавлены и другие алгоритмы и режимы работы, например опции для выравнивания некодирующих последовательностей РНК.
Алгоритм MAFFT работает следующим образом: попарное выравнивание, расчет расстояния, построение направляющего дерева, прогрессивное выравнивание, итеративное уточнение.
Попарное выравнивание: этот шаг используется для определения схожих областей между введенными последовательностями.
Направляющее дерево: с использованием матрицы расстояний строится направляющее дерево, в котором существует иерархическое представление кластеров (каждый узел является кластером), а включенные ветки представляют собой расстояние между кластерами.
Прогрессивное выравнивание: с помощью направляющего дерева прогрессивное выравнивание выполняется от листьев к корню.
Итеративное выравнивание. На этапе итеративного уточнения весь процесс повторяется с корректировкой положений промежутков и вставок для повышения точности выравнивания. Временная сложность итеративного выравнивания зависит от количества происходящих итераций.