Алгоритмы множественного выравнивания
Сравнение выравниваний, построенных разными программами
Для выравнивания были выбраны белки из семейства heparin-binding growth factors family, это семейство было выбрано по следующим критериям: чтобы последовательности были не очень длинными и можно было легко визуально проанализировать выравнивания, но и не слишком короткими, чтобы было, что анализировать; чтобы среди этих белков для многих была известна пространственная структура и была симпатичной для дальнейшего выравнивания по ней; чтобы сами белки были интересны с функциональной точки зрения. Для выравнивания я выбрала следующие 5 белков этого семейства:
| UniProt ID | Name | Organism | Function | PDB AC |
|---|---|---|---|---|
| FGF4_HUMAN | Fibroblast growth factor 4 | Homo sapiens | Регуляция эмбрионального развития, пролиферации и дифференцировки клеток | 1ijt |
| FGF2_HUMAN | Fibroblast growth factor 2 | Homo sapiens | Регуляция деления, миграций, дифференцировки, жизни клеток | 1bas |
| FGF12_HUMAN | Fibroblast growth factor 12 | Homo sapiens | Развитие и функционирование нервной системы | 1q1u |
| FGF7_RAT | Fibroblast growth factor 7 | Rattus norvegicus | Пролиферация клеток эпителия, фактор роста кератиноцитов | |
| FGF21_HUMAN | Fibroblast growth factor 21 | Homo sapiens | Поддержание гомеостаза глюкозы и чувствительности к инсулину |
Я сравнивала программу MUSCLE с программами T-Coffee и Clustal, проект JalView. Для сравения выравниваний использовалась программа Михаила Изгагина
Длины самих последовательностей отличаются вплоть до 90 аминокислот. Тем не менее они хорошо выровнялись на достоверном участке с координатам ~ 175-300 (MUSCLE). Длины выравниваний: MUSCLE: 362; T-Coffee: 374; Clustal: 334.
| MUSCLE | T-Coffee | block lenght | MUSCLE | Clustal | block lenght |
|---|---|---|---|---|---|
| 51-53 | 55-57 | 3 | 1-27 | 1-27 | 27 |
| 83-85 | 91-93 | 3 | 83-110 | 78-105 | 28 |
| 92-110 | 100-118 | 19 | 198-215 | 174-191 | 18 |
| 183-189 | 180-186 | 7 | 220-256 | 196-232 | 37 |
| 220-257 | 219-256 | 38 | 285-286 | 261-262 | 2 |
| 260-276 | 259-275 | 17 | |||
| 284-287 | 283-286 | 4 | |||
| 293-294 | 292-293 | 2 | |||
| 315-322 | 310-317 | 8 | |||
| 10 | 10 |
MUSCLE и Clustal имеют больший процент совпадающих колонок, чем MUSCLE и T-Coffee (35.64% против 32.87%), несмотря на то, что выравнивание Clustal примерно на 30 колонок короче, чем выравнивания MUSCLE и T-Coffee. MUSCLE и T-Coffee почти одинаковой длинны, но процент одинаковых колонок в них меньше, и выравнивания в целом отличаются больше, чем MUSCLE и Clustal. Также в сравнении MUSCLE и T-Coffee мы получаем много блоков, но большая часть из них короткая. В сравнении MUSCLE и Clustal блоков меньше, но они длиннее, что более правдоподобно с эволюционной точки зрения. То есть MUSCLE и Clustal справились с выравниванием лучше, чем T-Coffee. Выравние Clustal, видимо, имеет меньшую длинну за счет более оптимального выравнивания примерно с 1 по 160 колонку. По координатам одинаковых блоков также можно видеть, что консервативные блоки начинаются в выравнивании Clustal раньше, чем в выравниваниях MUSCLE и T-Coffee. Интересно сравнить Clustal со структурным выравниванием.
Сравнение со структурным выравниванием
Для структурного выравнивания были взяты первые три белка из выбранных выше, в таблице можно найти их PDB AC. Визуально структуры очень хорошо выравнялись вплоть до поворотов.
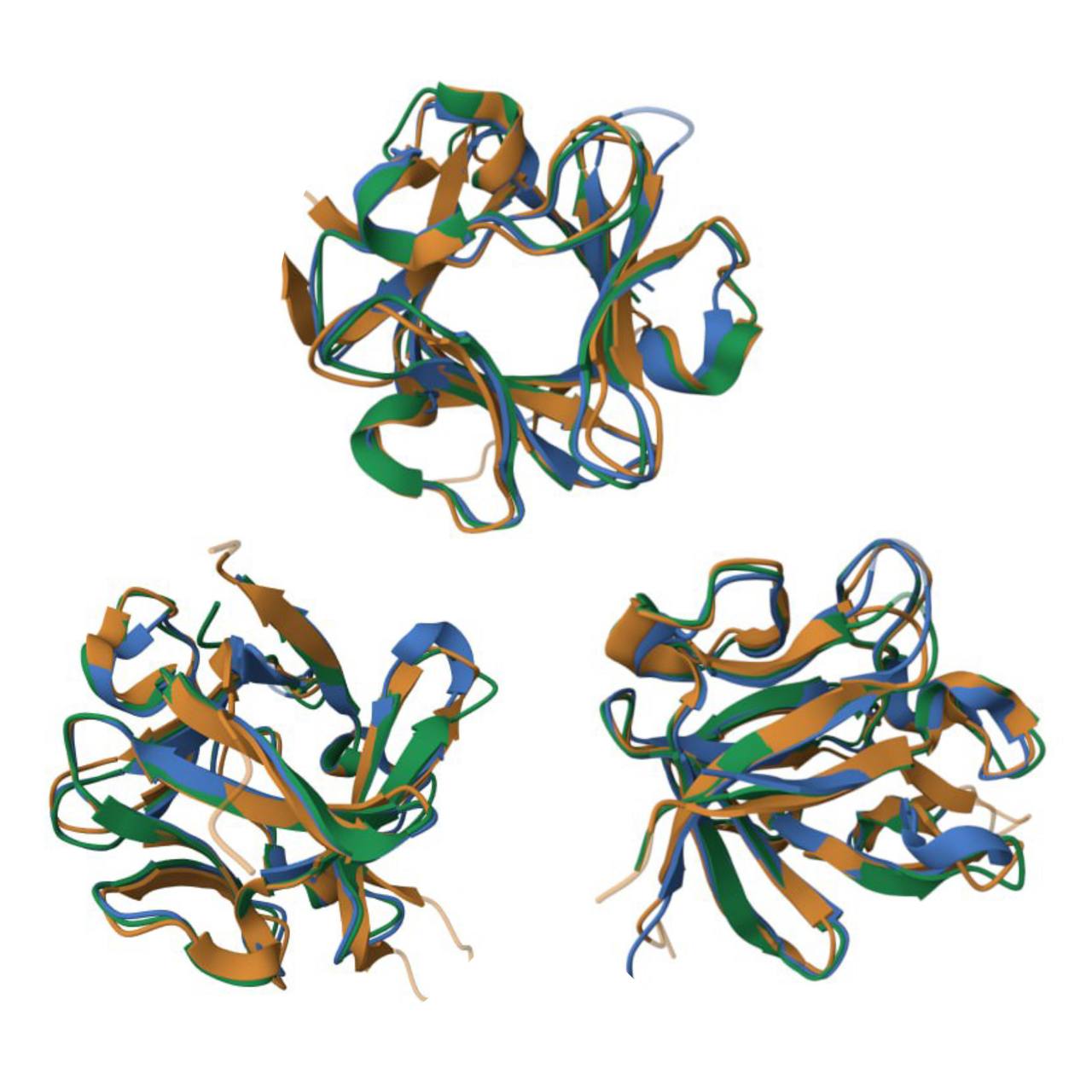
| Clustal | Structural alignment | block lenght |
|---|---|---|
| 11-16 | 9-14 | 6 |
| 20-36 | 18-34 | 17 |
| 40-138 | 38-136 | 99 |
| 141-142 | 139-140 | 2 |
| 18 | 16 |
Проект JalView. Сравнение выравниваний было выполнено программой Антона Куликова. Выравнивания одинаковой длинны и совпадают на 88%. То есть в данном случае алгоритмическое выравнивание показало очень точный результат относительно структурного. И на самом деле не только Clustle, но и MUSCLE и T-Coffee показали такой же хороший результат. Этого можно было ожидать, ведь участок последовательности достаточно консерввативен.
Описание работы алгоритма Clustal W
Алгоритм ClustalW использует прогрессивное выравнивание, то есть: попарно выравнивает последовательности, сторит матрицу растояний, строит напрявляющее дерево и, следуя ему, "достраивает" выравнивания для наиболее близких последовательностей, пока все последовательности не будут выравнены. Алгоритм отличается от остальных высокой чувствительностью, которая обеспечивается W - weighting — для последовательностей парных выравниваний расчитывается вес таким образом, что чем последовательности ближе эволюционно, тем он меньше, чем дальше — тем больше. Во-вторых, на каждой стадии выравнивания меняется матрица на наиболее подходящую в соответствии с дивергентностью последовательностей (т. е. на основе посчитанного веса). В-третьих, штрафы за гэп специфичны в соответствии с аминокислотными остатками, также снижается штраф за гэп при открытии гэпа на той же позиции, на которой он уже был открыт в другом выравнивании. Все это обеспечивает высокую точность алгоритма, в частности для нерегулярных участков.
Источники:
Using Clustal for multiple sequence alignment
CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment ...
Multiple Sequence Alignment Algorithms in Bioinformatics