Множественные выравнивания
Сравнивание выравниваний последовательностей разными программами
Для этого задания я взяла белки из практикума 9: HTPG_CLOBH, HTPG_BACSU, HTPG_HELPY, HTPG_ECOLI и HTPG_GEOSL. И провела их множественные выравнивания в Clustal Omega, T-Coffee, MUSCLE и MAFFT. Сравнила все пары выравниваний с помощью программы Лизы Плешко.
Было выявлено, что доля одинаково выравненных позиций в выравниваниях больше всего у пары MUSCLE и MAFFT (93% у каждого выравнивания) и меньше всего у пары Clustal Omega и MAFFT (85% у каждого выравнивания). Эти две пары я сравнила еще и в VerAlign. Результаты для первой пары: SP score: 0.97, CS score: 0.96, avg_SPdist score: 0.99. Для второй пары: SP score: 0.95, CS score: 0.93, avg_SPdist score: 0.99.
Проанализировав последовательности, можно заметить крупные участки наибольшего расхождения выравниваний – недостоверные участки – 218-242 и 510-525 (также есть более мелкие участки). Так как программы MSA работают по разным алгоритмам, то это значит, что есть большая вероятность того, что такие спорные участки будут выровнены по-разному.
Множественные выравнивания в Jalview
Выдача программы Лизы Плешко пары MUSCLE и MAFFT
Выдача программы Лизы Плешко пары Clustal Omega и MAFFT
VerAlign пары Clustal Omega и MAFFT
Сравнение выравнивания по совмещению структур с выравниванием MSA
Для этого задания я выбрала семейства доменов PNTB (PF02233) – бета-субъединица NAD(P) трансгидрогеназы. Для выравниваний я взяла домены из этого семейства из бактерии Rhodospirillum rubrum (PDB_ID: 1E3T), быка (PDB_ID: 1D4O) и человека (PDB_ID: 1DJL).
Выравнивание структур было произведено в PyMOL (align). На картинках бирюзовым цветом обозначен домен из бактерии, зеленым – из быка и розовым – из человека. Как видно из выравнивания домены быка и человека более схожи между собой, чем с бактериальным доменом (что не удивительно). Выравнивание последовательностей с помощью Clustal Omega подтверждает это.
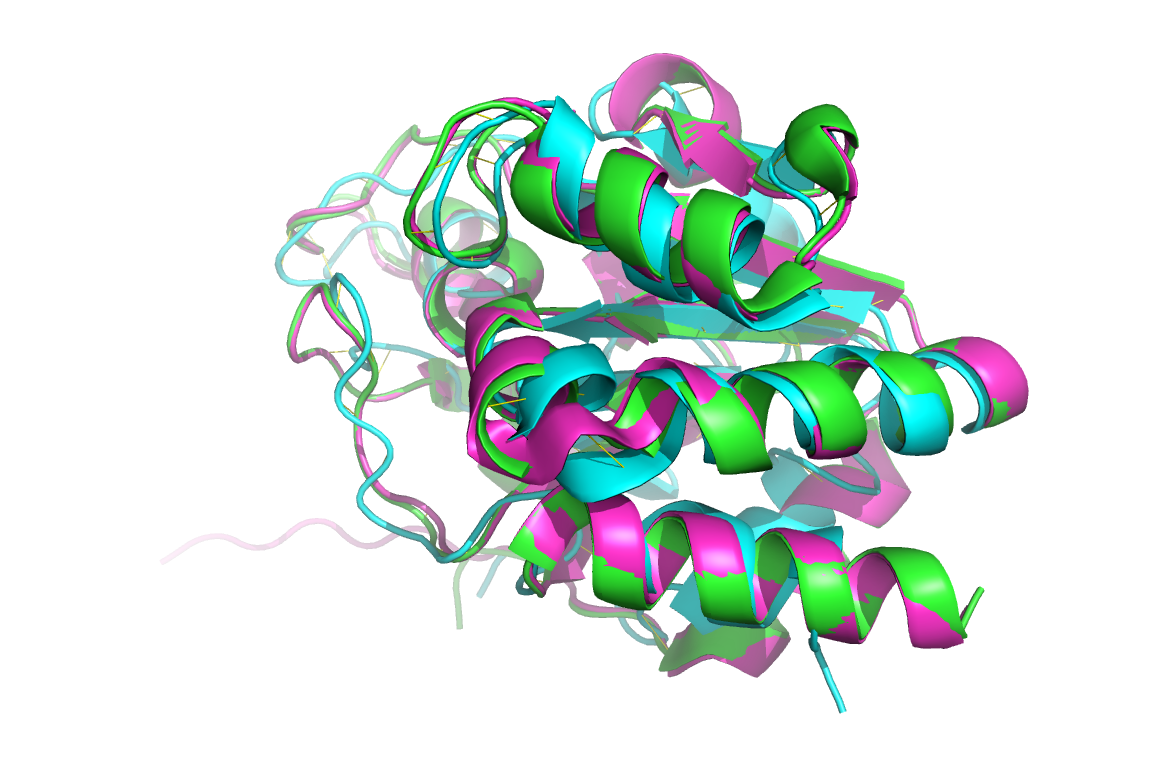
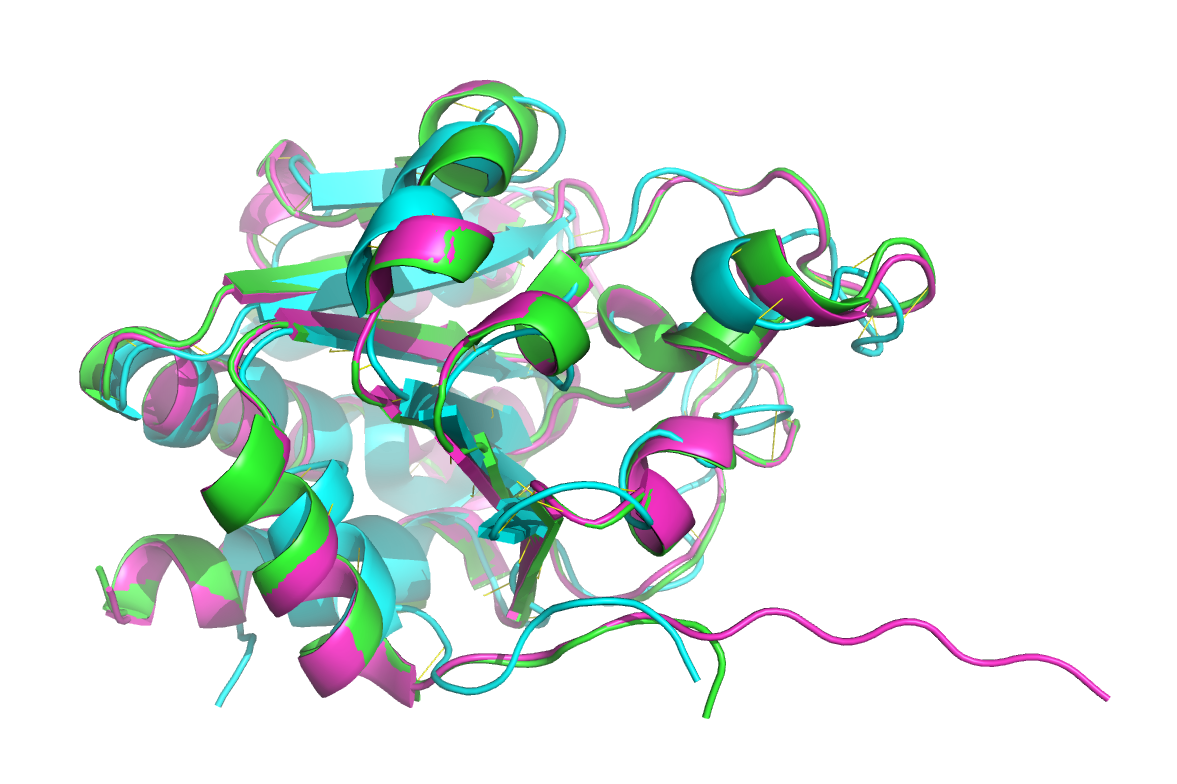
Множественное выравнивание с помощью Clustal Omega в Jalview
Описание программы MAFFT
MAFFT – это программа множественного выравнивания, использующая быстрое преобразование Фурье.
Опубликованная в 2002 году первая версия MAFFT использовала алгоритм, основанный на прогрессивном выравнивании, в котором последовательности группировались с помощью быстрого преобразования Фурье. В последующих версиях MAFFT были добавлены другие алгоритмы и режимы работы, включая варианты более быстрого выравнивания большого количества последовательностей, более точное выравнивание, выравнивание некодирующих последовательностей РНК.
Алгоритм MAFFT работает, следуя 5 шагам: попарное выравнивание, расчет расстояния, построение направляющего дерева, прогрессивное выравнивание, итеративное уточнение. Описание работы:
Используя рассчитанные попарные выравнивания, выполняется вычисление матрицы расстояний для оценки различий между выравниваниями. По данным матрицы расстояний строится направляющее дерево, в котором имеется иерархическое представление кластеров последовательностей. С помощью направляющего дерева выполняется прогрессивное выравнивание от листьев к корню. Алгоритм использует введенные последовательности и выравнивает дочерние узлы, чтобы вычислить согласованное выравнивание для родительского узла. Этот шаг выполняется до тех пор, пока не будет пройдено все дерево, что приведет к окончательному выравниванию множественных последовательностей. Итеративный этап уточнения повторяет весь процесс с корректировкой положения гэпов для повышения точности выравнивания.