| Шаг | Команда | Дополнительно |
| Анализ качества данных однонуклеотидных чтений | >fastqc chr11_2.fastq | Результат доступен по ссылке: FastQC11_2-report |
| Очистка чтений (качество не ниже 20 с конца кажого чтения + ограничения по длине) | >java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr11_1.fastq trimmed11_1.fastq LEADING:20 TRAILING:20 MINLEN:50 | Осталось 33893, вырезано 357 ридов |
| Анализ качества очищенных чтений | >fastqc trimmed11_2.fastq | Результат доступен по ссылке: FastQC11_2_trimmed-report |
Сравнение результатов до и после очистки некачественных ридов. Из рис. 1 и 2 видно, что качество ридов было хорошоим и до очитки, так как риды не выходили за пределы зеленой зоны, после очистки качество всё же немного улучшилось, поэтому будем использовать улучшенный файл.
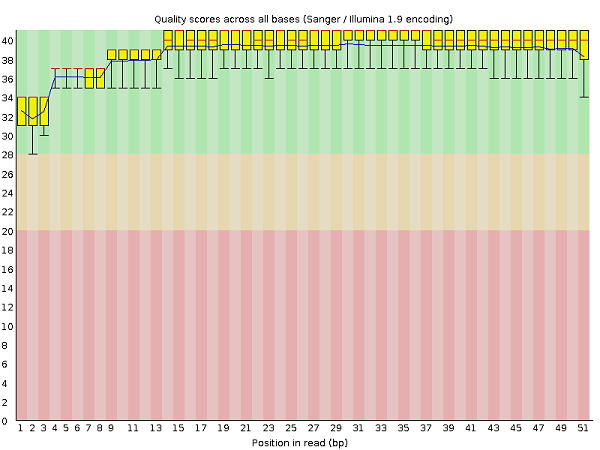
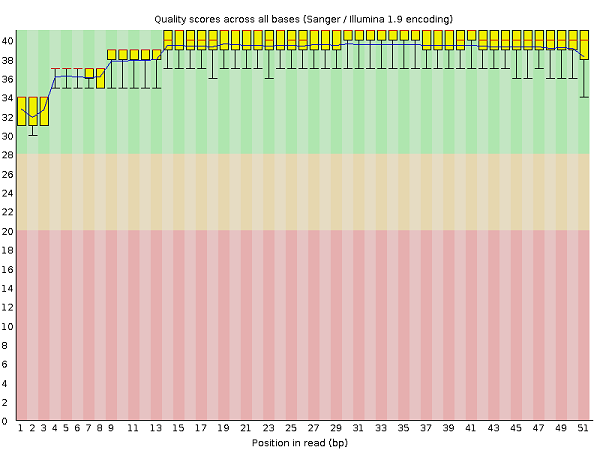
Рис.1 и 2 "Per base sequence quality" до и после обработки ридов в программе Trimmomatic.
Картирование чтений включало в себя индексацию референсной последоваельности и построение выравниваний прочтений и референса в формате .sam.
>Пусть к программам, необходимым для запуска Hisat2: export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
>Индексирование референса: hisat2-build chr11.fasta chr11 (*)
>Построение выравниваний: hisat2 -x chr11 -U trimmed11_2.fastq -S align11_2.sam --no-softclip(**)
>Индексирование референса: hisat2-build chr11.fasta chr11 (*)
>Построение выравниваний: hisat2 -x chr11 -U trimmed11_2.fastq -S align11_2.sam --no-softclip(**)
* - подробнее об этом шаге можно прочитать в отчете о выполнении практикума 11: pr11. В частности, для выполнения этой команды использовались те же индексные файлы.
** - в отличие от практикума 11, в этом случае не использовался параметр "--no-spliced-alignmet": при анализе транскриптомов нужно учитывать сплайсинг, поэтому нужно разрешить разрывы.
Анализ выравнивания
Были последовательно выполнены следующие команды:
| Команда | Функция |
| samtools view -b align11_2.sam -o align11_2.bam | Переводит выравнивание (референс + чтения) из формата .sam в бинарный формат .bam |
| samtools sort align11_2.bam -T align11_2.txt -o align11_2_sorted.bam | Сортирует выравнивание по коортинате референса в начале чтения |
| samtools index align11_2_sorted.bam | Индексирует полученный после сортировки .bam-файл |
| samtools idxstats align11_2_sorted.bam >idxstats11_2.txt | Подсчитывет количество чтений, откартированных на геном и выводит результат в файл idxstats11_2.txt |
В результате проведенных опрераций 33625 чтений картировались на геном, 268 - нет.
Подсчёт чтений с помощью Bedtools
Для того чтобы выяснить какие гены и насколько глубоко оказались покрыты чтениями, я вырлнила команды, представленные в таблице ниже.
| Команда | Функция |
| /P/y14/term3/block4/SNP/bedtools2/bin/bedtools bamtobed -i align11_2_sorted.bam > reads11_2.bed | .bam файл переводится в .bed файл (в него кладутся координаты выровненных ридов) |
| /P/y14/term3/block4/SNP/bedtools2/bin/bedtools intersect -a /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed -b reads11_2.bed -c > overlap11_2.bed | На выходе получаем файл .bam формата с пересечением разметки генов с получееными координатами |
| sort -k 6 -r overlap11_2.bed > sort_overlap11_2.bed | Сортировка файла, полученного в предыдущем шаге |
Проанализировав выходной файл sort_overlap11_2.bed, я пришла в выводу, что мои прочтения в оновном попали в границы гена HSPA8, а также snoРНК SNORD14D и SNORD14С. HSPA8 - это ген белка теплового шока "heat shock protein family A (Hsp70) member 8", а snoРНК - это малые ядрошковые РНК, класс малых РНК, которые обычно участвуют в метилировании и псевдоудилировании рибосомных РНК. В моем случае это SNORD14D и SNORD14С snoРНК.
Упражнения по bedtools
Задание 1. Получение файла с ридами (формат fastq) из файла с выравниваниями.
>/P/y14/term3/block4/SNP/bedtools2/bin/bedtools bamtofastq -i align11_2_sorted.bam -fq reads_from_align.fq
>На выходе получаю файл: reads_from_align.fq
>/P/y14/term3/block4/SNP/bedtools2/bin/bedtools bamtofastq -i align11_2_sorted.bam -fq reads_from_align.fq
>На выходе получаю файл: reads_from_align.fq
Задание 4. Объединение моих чтений в кластеры - было получено 30 кластеров!
>/P/y14/term3/block4/SNP/bedtools2/bin/bedtools cluster -i reads11_2.bed > clusters11_2.bed
>На выходе: clusters11_2.bed
>/P/y14/term3/block4/SNP/bedtools2/bin/bedtools cluster -i reads11_2.bed > clusters11_2.bed
>На выходе: clusters11_2.bed
Задание 2. Получение файла с нуклеотидной последовательностью (.fasta) для одного из покрытых чтениями генов.
>/P/y14/term3/block4/SNP/bedtools2/bin/bedtools getfasta -fi chr11.fasta -bed reads11_2.bed > nucl11_2.fasta
>Выходный файл: nucl11_2.fasta
>/P/y14/term3/block4/SNP/bedtools2/bin/bedtools getfasta -fi chr11.fasta -bed reads11_2.bed > nucl11_2.fasta
>Выходный файл: nucl11_2.fasta