Задание 3
Для создания базы данных ,sk использован файл генома S. cerevisiae (сборка R64, GCF_000146045.2_R64_genomic.fna), скачанный с NCBI.
Индексация выполнена командой:
makeblastdb -in S_cerevisiae_genome.fasta -dbtype nucl -out S_cerevisiae_db -parse_seqids
Разбор команды
Команда makeblastdb используется для создания локальной базы данных BLAST из файла с последовательностями (обычно в формате FASTA). Эта база данных затем используется программами BLAST (например, blastn, blastp, tblastn) для поиска гомологичных последовательностей.
-in S_cerevisiae_genome.fasta
Указывает входной файл, содержащий последовательности в формате FASTA. В данном случае это полный геном дрожжей Saccharomyces cerevisiae (хромосомы, митохондриальная ДНК, плазмиды). Файл должен быть в текстовом формате, каждая запись начинается с символа .
-dbtype nucl
Определяет тип базы данных:
nucl — нуклеотидная база (для программ, работающих с ДНК/РНК, например blastn, tblastn).
prot — белковая база (для blastp, blastx).
-out S_cerevisiae_db
Задаёт префикс (базовое имя) для выходных файлов базы данных. В результате будут созданы несколько файлов с этим префиксом и различными расширениями (например, S_cerevisiae_db.nhr, S_cerevisiae_db.nin, S_cerevisiae_db.nsq). Эти файлы содержат индексы и последовательности в специальном формате, оптимизированном для быстрого поиска BLAST.
-parse_seqids
Эта опция указывает программе извлечь идентификаторы последовательностей из заголовков FASTA (строк, начинающихся с больших). Без этой опции BLAST будет использовать автоматически сгенерированные идентификаторы (например, lcl|1). С опцией -parse_seqids в результатах BLAST будут отображаться исходные идентификаторы (например, NC_001144.5), что удобно для интерпретации. Опция необходима, если вы планируете использовать -seqidlist или хотите обращаться к последовательностям по их оригинальным ID.
После успешного выполнения команды в текущем каталоге появятся файлы:
- S_cerevisiae_db.nhr (заголовок базы)
- S_cerevisiae_db.nin (информация о последовательностях)
- S_cerevisiae_db.nsq (сами последовательности)
- возможно, другие вспомогательные файлы (.nog, .nos, .not, .ndb и т.д.)
Далее был запущен алгоритм blastn (т.к. подходит для поиска похожих некодирующих нуклеотидных последовательностей даже среди неблизкородственных организмов)
blastn -task blastn -query Ecoli_16S.fasta -db S_cerevisiae_db -out r16 -outfmt 7
blastn -task blastn -query Ecoli_16S.fasta -db S_cerevisiae_db -out r16.txt -outfmt 0
blastn -task blastn -query Ecoli_23S.fasta -db S_cerevisiae_db -out r23 -outfmt 7
blastn -task blastn -query Ecoli_23S.fasta -db S_cerevisiae_db -out r23.txt -outfmt 0
Колонки и разбор кода
- query acc. — идентификатор запроса (например, NR_103073.1).
- subject acc. — идентификатор последовательности в базе (хромосома или контиг).
- % identity — процент идентичности.
- alignment length — длина выравнивания.
- mismatches — число несовпадений.
- gap opens — число открытых гэпов.
- q. start — начало на запросе.
- q. end — конец на запросе.
- s. start — начало на целевой последовательности.
- s. end — конец на целевой последовательности.
- evalue — ожидаемое значение (статистическая значимость).
- bit score — битовый скор.
Разбор команды
Команда blastn используется для поиска нуклеотидных последовательностей-запросов в нуклеотидной базе данных. Она сравнивает заданную последовательность со всеми последовательностями в подготовленной базе BLAST и выдаёт участки сходства (хиты). В данном случае выполняется классический нуклеотидный BLAST (task blastn) с запросом в виде 16S рРНК E. coli против базы данных генома дрожжей S. cerevisiae.
-task blastn
Задаёт вариант алгоритма BLAST для нуклеотидов.
-query Ecoli_16S.fast
Указывает путь к файлу с последовательностью-запросом (или несколькими). Файл должен быть в формате FASTA (обычно расширения .fasta, .fa, .fast и т.п.).
-db S_cerevisiae_db
Имя базы данных, с которой будет выполняться сравнение. Это префикс, заданный при создании базы данных с помощью makeblastdb. В данном случае база называется S_cerevisiae_db и, судя по имени, содержит нуклеотидные последовательности генома дрожжей Saccharomyces cerevisiae. Программа автоматически найдёт файлы с расширениями .nhr, .nin, .nsq и др., соответствующие этой базе.
-out res16_2
Имя файла, в который будут записаны результаты поиска. Если не указать этот параметр, вывод направляется в стандартный поток (на экран). Здесь результаты сохраняются в файл res16_2.
-outfmt 0
— стандартный «парный» вывод с подробным описанием каждого выравнивания (что удобно для просмотра вручную).
-outfmt 7
— табличный формат с комментариями, удобен для импорта в электронные таблицы и автоматической обработки.
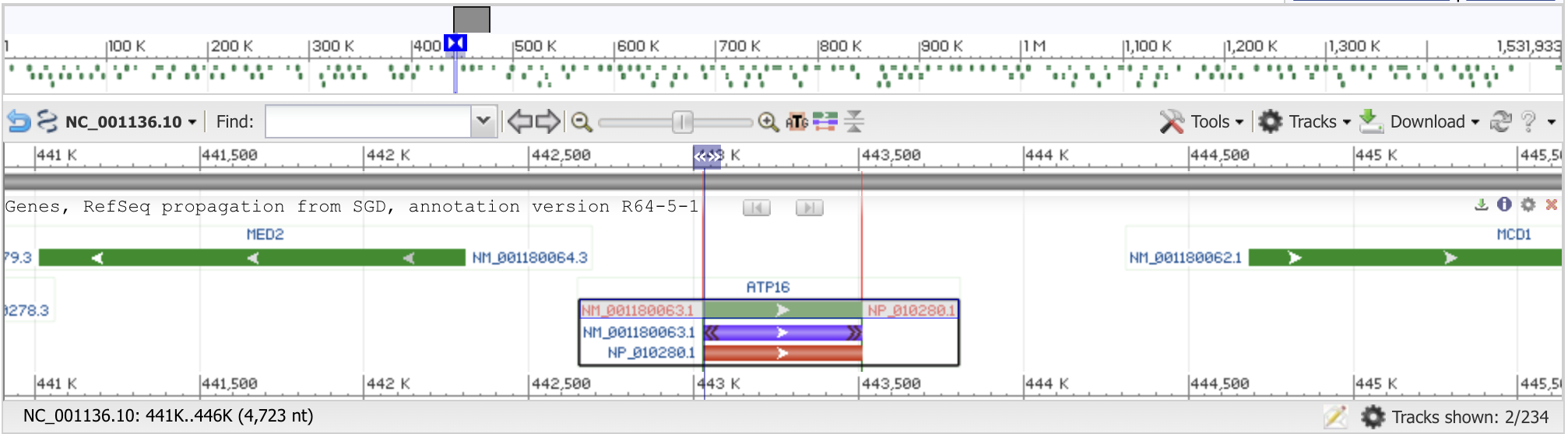
У дрожжей Saccharomyces cerevisiae (грибы) есть два типа рибосом:
- Цитоплазматические рибосомы (80S), состоящие из малой (40S) и большой (60S) субъединиц. В их состав входят рРНК: 18S (малая субъединица), 5.8S, 25S и 5S (большая субъединица).
- Митохондриальные рибосомы (70S-подобные), которые обеспечивают трансляцию внутри митохондрий. Они содержат 15S рРНК (аналог 16S) и 21S рРНК (аналог 23S), а также, возможно, 5S рРНК (у некоторых видов, но у дрожжей их нет).
Значит, гомологи 16S рРНК E. coli у дрожжей должны соответствовать 18S рРНК (цитоплазматическая) и 15S рРНК (митохондриальная). Гомологи 23S — 25S рРНК (цитоплазматическая) и 21S рРНК (митохондриальная). Гомологи 5S — цитоплазматическая 5S рРНК.
Результаты выдачи:
Соответсвенно, среди гомологов 16S рРНК E. coli у дрожжей нашлись 2 выравнивания на хромосоме XII (NC_001144.5) соответствуют разным копиям гена 18S рРНК ( Score ~56.3 и E-value = 3.94e-07). На митохондриальном геноме (NC_001224.1) обнаружены два участка, относящихся к гену 15S рРНК. Таким образом, число уникальных гомологов для 16S рРНК равно двум: ядерный ген 18S рРНК и митохондриальный ген 15S рРНК, что полностью соответствует ожиданиям.
Среди гомологов 23S рРНК E. coli все значимые выравнивания приходятся на две геномные мишени: митохондриальную ДНК (NC_001224.1) и хромосому XII (NC_001144.5). На митохондриальной ДНК обнаружены два длинных выравнивания (187 и 171 п.н.), соответствующих гену 21S рРНК – гомологу бактериальной 23S рРНК в митохондриальной рибосоме. На хромосоме XII было найдено множество выравниваний , которые представляют собой различные копии гена 25S рРНК. Таким образом, число уникальных гомологов для 23S рРНК равно двум: митохондриальный ген 21S рРНК и ядерный ген 25S рРНК. Этот результат полностью согласуется с теоретическими ожиданиями.
Задание 4
Путём тыканья пальцем в небо, мной был выбран род Yersinia, относящийся к семейству Yersiniaceae и включающий грамотрицательные палочковидные бактерии
Пара нуклеотидных последовательностей:
Поиск исходных геномных сборок производился в базе данных "Nucleotide" NCBI с использованием запроса: "Yersinia"[Organism] AND "complete genome"[Title]
Сравнение последовательностей осуществлялось при помощи алгоритмов megablast и blastn со стандартными настройками. Для поиска megablast доля покрытия запроса составила 83%, а уровень идентичности нуклеотидных остатков — 89.11%. При применении стандартного blastn аналогичные параметры оказались равны 85% и 89.03% соответственно. Наблюдаемые цифры вполне ожидаемы, поскольку сопоставлялись геномы двух представителей одного рода. Кроме того, полученные данные предварительно указывают на то, что blastn способен детектировать более тонкие различия, нежели megablast (это предположение впоследствии подтвердилось при визуальном анализе графиков локального сходства).
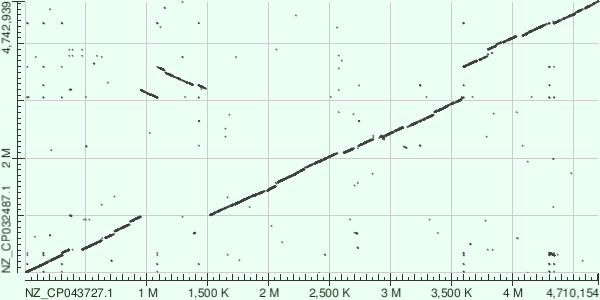
Диаграмма локальных совпадений между хромосомами Yersinia hibernica CFS1934 и Yersinia canariae NCTC 14382, построенная с применением megablast
Исходя из графка, можно предположить, что произошла серия из двух инверcий. Пocле первoй изменилacь oриентaция центрaльнoгo учaсткa, пoсле втoрoй произошло чвстичное востановление. Также после инверсий заметна транслокация, такде в центральной части. А по углам можно заметить делеции/инсерции, на это нам указывает характерный "обрыв" прямых.
На следующем этапе для построения карты был задействован исключительно алгоритм blastn без подключения megablast. Результирующее изображение представлено ниже:
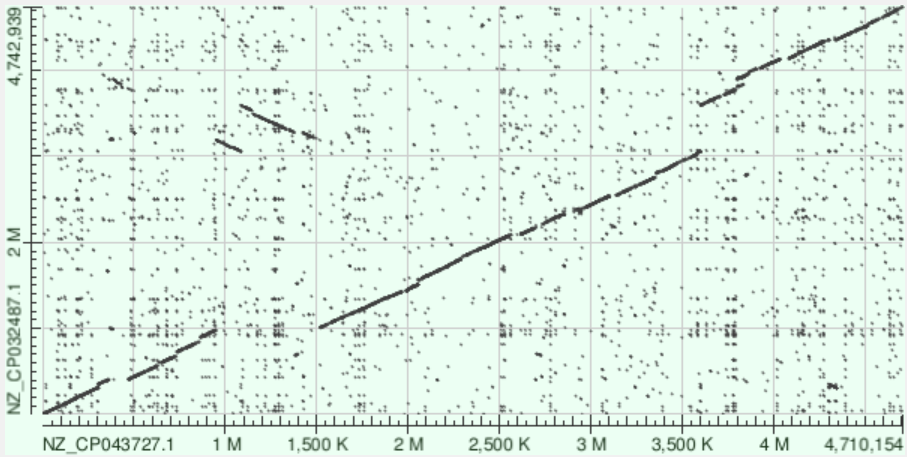
Диаграмма локальных совпадений между хромосомами Yersinia hibernica CFS1934 и Yersinia canariae NCTC 14382, построенная исключительно с помощью blastn
Общая картина распределения сигналов оказалась весьма сходной с предыдущей (что закономерно). Тем не менее, на данной диаграмме присутствиет фонового рассеяния — множественных коротких совпадений, распознанных blastn, но невидимых для megablast.
Резюмируя , можно с уверенностью утверждать, что поисковый метод blastn более чувствительный по сравнению с megablast.