Сравнение матриц аминокислотных замен
Зачем нужны матрицы замен? Они необходимы для оценки качества выравнивания аминокислотных (или нуклеотидных) последовательностей. В такой матрице каждая аминокислота сопостовляется каждой, и все подобные пары получают некоторый вес, который зависит от частоты встречаемости такой замены (чем чаще - тем больше). Выравнивание тем лучше, чем больше сумма весов всех замен. существуют разные виды матриц, например, BLOSUM и PHAT.
Откуда берутся матрицы замен? Матрица BLOSUM строится на основе базы данных BLOCKS, которая содержит блоки выравниваний множества белков из различных семейств. В этих выравниваниях подсчитывается число замен каждого типа. Затем находят наблюдаемые и ожидаемые частоты встречаемости каждой возможной пары аминокислот. Чем больше отношение этих величин, тем больше вес замены.
Матрица PHAT строится несколько иначе. В качестве исходных данных для матрицы наблюдаемых частот используются блоки, которые по предсказанию программы PHDhtm являются трансмембранными. Для матрицы ожидаемых частот используются блоки, в которых высоко содержание гидрофобных аминокислот (Ng P.C., et al. (2000) PHAT: a transmembrane-specific substitution matrix. Predicted hydrophobic and transmembrane. Bioinformatics). Значения в матрице PHAT отличаются от значений в BLOSUM, потому что в мембранных белках частоты встречаемости аминокислот другие.
Что за числа приписываются в конце матрицы? Если две последовательности имеют высокий процент идентичности, частоты пар аминокислот ошибочно изменяются. Чтобы избежать искажения результатов, такие последовательности объединяют в кластеры. Число 62 в названии матрицы - минимальный процент идентичности, при котором последовательности объединяются в кластер, который обрабатывается, как единое целое.
В чём проявляются различия?Для примера рассмотрим вес замен остатка лизина на другие остатки в разных матрицах (таблица 1). Значения из BLOSUM62 и из матрицы, построенной мной (промежуточные таблицы, использованные для постороения, можно найти в этом файле), различаются, так как база данных BLOCKS уже обновлялась и в качестве исходных были использованы немного другие частоты. Значения матрицы PHAT сильно отличаются и от BLOSUM62, и от моей, так как частоты встречаемости аминокислот в цитоплазматических и мембранных белках разные. Примерно одинаковым (причём положительным) сохраняется только вес замены остатка лизина на самого себя, так как по сути это не замена, а сохранение остатка, что часто наблюдается в эволюции белков.
| Таблица 1. |
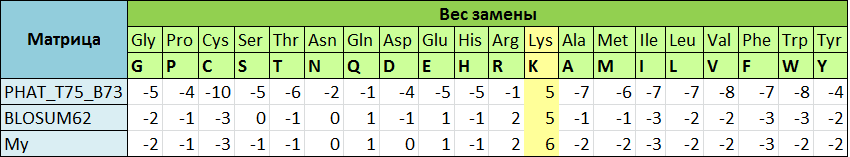
|
Сравнение выравниваний, полученных для коротких мутантов вручную и построенных классическими алгоритмами Нидлмана-Вунша и Смита-Ватермана
Выравнивание двух последовательностей можно осуществить вручную. Это долго и неудобно, особенно, если последовательности достаточно длинные. Поэтому были придуманы алгоритмы, которые могут выполнять эту работу.
Программа needle из пакета EMBOSS использует алгоритм глобального выравнивания Нидлмана-Вунша. Исходные данные - две последовательности, которые нужно выровнять. Настраиваемые параметры программы:
- datafile - матрица весов аминокислотных или нуклеотидных замен, по умолчанию BLOSUM62 или DNAfull соответственно,
- gapopen - штраф за открытие гэпа (вес пропуска, который следует за аминокислотой), по умолчанию равен 10,
- gapextend - штраф за продолжение гэпа (вес пропуска, который следует за другим пропуском), по умолчанию равен 0,5.
- endopen - штраф за открытие гэпа (вес пропуска, который следует за аминокислотой), по умолчанию равен 10,
- endextend - штраф за продолжение гэпа (вес пропуска, который следует за другим пропуском), по умолчанию равен 0,5.
Программа needle находит выравнивание (по сути - расстановку гэпов), при которой вес выравнивания максимальный. При этом пропуски, установленные в начале или конце последовательности, штрафуются.
Программа water из пакета EMBOSS использует алгоритм локального выравнивания Смита-Ватермана. Исходные данные те же, но из настраиваемых параметров остаются только datafile, gapopen и gapextend, гэпы в конце и в начале последовательности не штрафуются. Это главное отличие от алгоритма Нидлмана-Вунша. water найдёт лучшее выравнивание короткой последовательности с более длинной, без появления лишних гэпов посреди последовательности. needle найдёт лучшее выравнивание двух последовательностей примерно одинакового размера.
В чём проявляются различия? Для примера сравним выравнивания вручную, с помощью needle и с помошью water
| Мутант | Ручное выравнивание | Выравниевание needle | Выравнивание water |
| 1 | 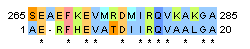
|

|

|
| Identity: 13/20 ( 57%) Similarity: 14/20 ( 67%) Gaps: 1/20 (5%) Score: 50 |
Identity: 13/383 ( 3.4%) Similarity: 14/383 ( 3.7%) Gaps: 364/383 (95.0%) Score: 47.0 |
Identity: 11/17 ( 64.7%) Similarity: 12/17 ( 70.6%) Gaps: 0/17 (0.0%) Score: 48.0 |
|
| 2 | 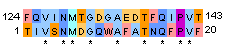
|

|

|
| Identity: 9/20 ( 45%) Similarity: 11/20 ( 55%) Gaps: 0/20 (0%) Score: 26 |
Identity: 9/382 ( 2.4%) Similarity: 9/383 ( 2.4%) Gaps: 362/382 (94.8%) Score: 26.0 |
Identity: 9/17 ( 52.9%) Similarity: 9/17 ( 52.9%) Gaps: 0/17 (0.0%) Score: 33.0 |
|
| 3 | 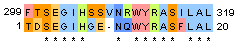
|

|

|
| Identity: 14/20 ( 67%) Similarity: 16/20 ( 76%) Gaps: 1/20 (5%) Score: 70 |
Identity: 16/382 ( 4.2%) Similarity: 17/383 ( 4.5%) Gaps: 362/382 (94.8%) Score: 67.5 |
Identity: 16/23 ( 69.6%) Similarity: 17/23 ( 73.9%) Gaps: 3/23 (13.0%) Score: 67.5 |
Во всех трёх случаях выравнивание water даёт больший вес. Это связано в основном с тем, что needle считает вес для всей последовательности мутаната, а water "обрезает" концы, если там нет совпадений. Кроме того, идентичность и сходство сравнимы только у ручных выравниваний и выравниваний water, так как needle считает процент от большей последовательности, а не от выровненного участка.