Эволюционные домены
Выбор домена
Для работы был выбран домен SNARE - (soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein (SNAP) receptors). Этот белок служит для обеспечения направления везикулярного транспорта у эукариот. Данные о домене приведены в таблице 1.
Таблица 1.
| АС | PF05739 |
| ID | SNARE |
| Число архитектур | 51 |
| Число последовательностей | 4036 |
| Число видов | 377 |
В JalView получим выравние всех последовательностей, содержащих домен. В меню File выберем пункт Fetch sequences, выберем базу данных PFAM, укажем необходимый AC. Выравнивание было окрашено по консервативности (ClustalX, порог - 10%). К одной из последовательностей прикрепили структуру из PDB (последовательность - SSOI_YEAST, PDB-код - 1fio). Были сохранены два файла - выравнивание в формате fasta и проект JalView в формате jar.
Выбор архитектур
Для дальнейшей работы были выбраны две архитектуры, в которые входит домен SNARE. Данные о них приведены в таблице 2.
Таблица 1.
| Домены | Изображение | Число последовательностей | Описание состава архитектуры |
| SNARE (PF05739) | 
|
1412 | C-концевой домен белка SNARE (домен SNARE и белок SNARE - разные вещи). Отвечает за закрепление комплекса в мембране, также может отвечать за распознавание органеллы-мишени, в которую направляется везикула. |
| Syntaxin (PF00804), SNARE (PF05739) | 
|
1370 | C-концевой домен белка SNARE + N-концевой домен этого белка - синтаксин. Синтаксин связывается со специфическим комплексом на мембране органеллы-мишени и обеспечивает распознавание. |
Исходя из данных сайта PFAM о распространении домена в различных таксономических группах, выберем таксон и подтаксоны, последовательности из которых будем сравнивать. Удобно взять в качестве таксона домен Eukaryota, подтаксоны - Fungi, Viridiplantae и Metazoa. Теперь нужно сделать выборку по нескольку белков из каждого подтаксона для каждой архитектуры.
Получим таблицу с информацией о последовательностях, содержащих изучаемый домен. Для этого воспользуемся скриптом swisspfam_to_xls.py, запустив его следующей командой:
python swisspfam_to_xls.py -z swisspfam.gz -p PF005739 -o snare.xlsПараметры -z, -p и -о задают входной архивированный файл, PFAM АС и выходной файл соответственно.
Теперь добавим в таблицу данные о таксономической принадлежности белков. Из полученной таблицы возьмём столбец с АС последовательностей в Uniprot и получим на
сайте этой базы данных записи обо всех последовательностях с помощью функции Retrieve. Оказалось, некоторые записи были изменены, пришлось изменить их и в нашей таблице,
иначе потом были бы несоответствия с полученной таксономией. Теперь сохраним flat text в отдельный файл, затем выберем из него только информацию о таксономии
с помощью скрипта uniprot_to_taxonomy.py и следующей команды:
После совмещения данных двух таблиц, построим сводную таблицу о принадлежности всех доменов ко всем последовательностям, и с её помощью отберём несколько белков, содержащие выбранные архитектуры и принадлежащих к выбранным таксонам. Таблицу со всеми вышеперечисленными данными можно скачать по этой ссылке.
Построение филогенетического дерева
Названия последовательностей были изменены - в начале к ним приписали код: буква V, F или М для обозначения Viridiplantae, Fungi или Metazoa соответственно, и цифра - 1 (SNARE) или 2 (SNARE + Syntaxin), соответствующая архитектуре. Для последовательностей белков из выборки было построено выравнивание с помощью программы Muscle. Выравнивание было окрашено методом ClustalX с порогом консервативности 5%. Выравние ришлось отредактировать - удалить плохо выровненные концы последовательностей, а также удалить несколько последовательностей, которые имели гепы в консервативных блоках, из-за чего выборка несколько сократилась. Итог на рисунке 1, или в сохранённом проекте, или в формате fasta.
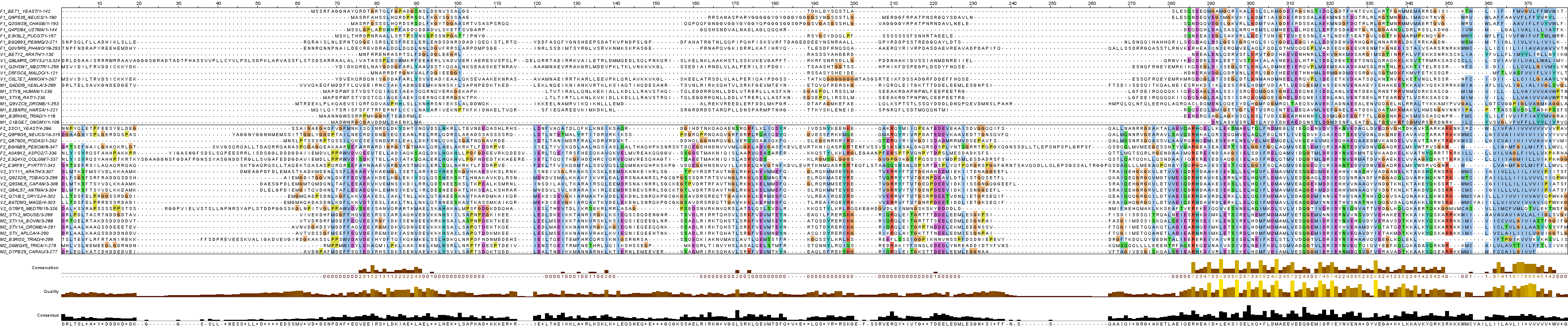
|
| Рис. 1. Отредактированное выравнивание последовательностей белков из выборки. |
По полученному выравниванию с помощью программы Mega было построено филогенетическое дерево методом Minimum evolution с бутстреп оценкой (см. рис. 2). Дерево было сохранено в формате nwk, и затем было получено другое отображение дерева с окрашенными ветвями с помощью программы iTOL (см. рис. 3).
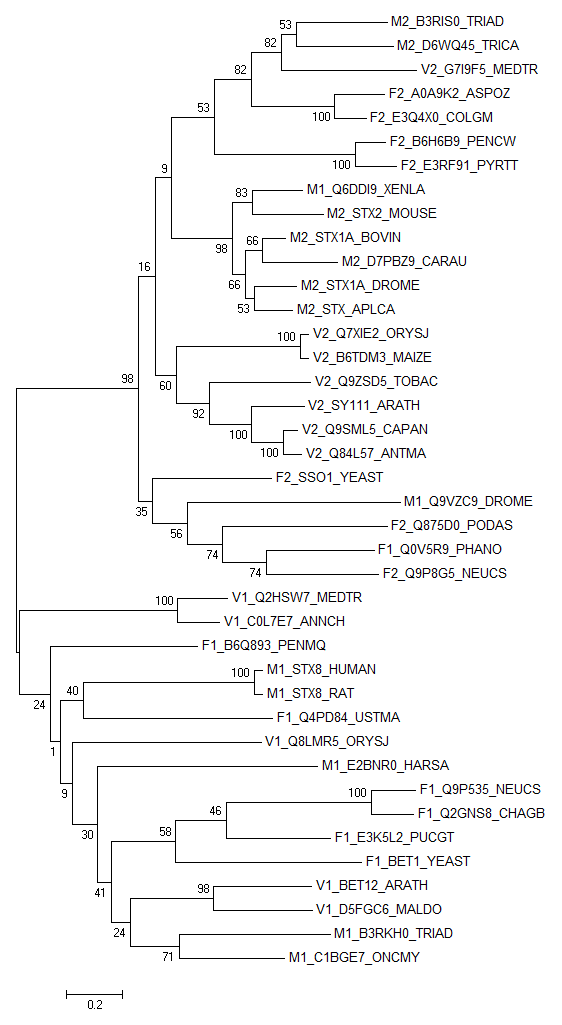
|
| Рис. 2. Дерево, полученное программой Mega. У каждой ветви подписан индекс поддержки. |
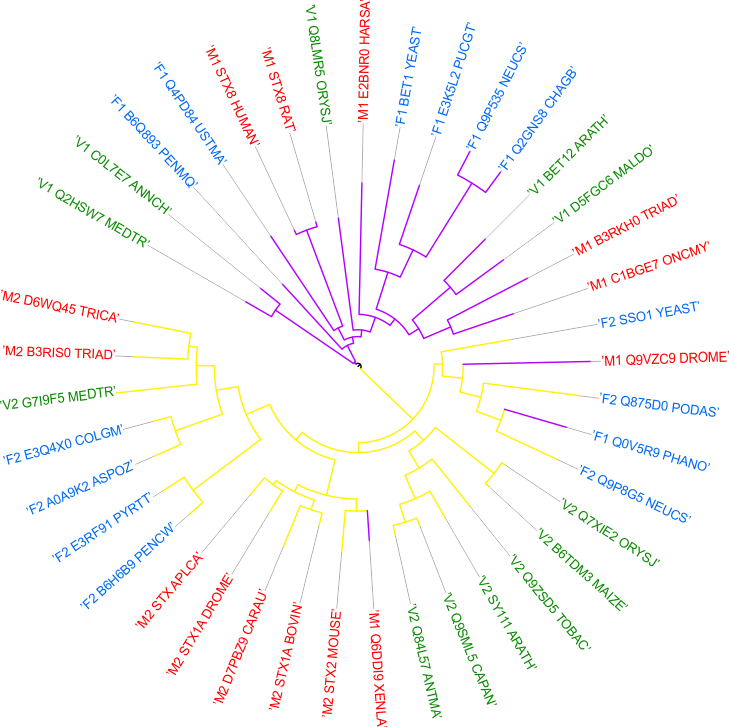
|
| Рис. 3. Дерево, построенное программой iTOL. Ветви, окрашенные фиолетовым соответствуют архитектуре SNARE, жёлтым - SNARE+Syntaxin. Названия последовательностей окрашены в соответствии с таксонами - зелёный для Viridiplantae, синий для Fungi и красный для Metazoa. |
Выводы
На раскрашенном дереве хорошо видно, что сначала разделяются архитектуры, а затем, внутри ветвей, соответствующих конкретной архитектуре - таксоны. Значит, обе архитектуры присутствовали уже у общего предка растений, животных и грибов. Наблюдается несколько выбросов - M1_Q9VZC9_DROME, F1_Q0V5R9_PHANO и M1_Q6DDI9_XENLA, скорее всего это связано с не очень хорошим выравниванием этих последовательностей. На выравнивании можно увидеть, что в консервативном блоке своей группы некоторые позиции этих последовательностей отличаются от консервативных.
Внутри ветви, соответствующей архитектуре SNARE+Syntaxin, листья, принадлежащие одному подтаксону, чаще расположоны в одной кладе, тогда как на ветви другой архитектуры листья различных таксонов расположены вперемешку. Вероятно, подтаксоны разошлись незадолго после того, как появилась архитектура SNARE+Syntaxin, после чего она эволюционировала в каждом подтаксоне отдельно. В внутри архитектуры SNARE эволюционные пути трудно проследить.
Стоит отметить, что не все ветви имеют хорошую поддержку. Особенно низка поддержка у ветвей, которые содержат много листьев, куда входят обе архитектуры или разные подтаксоны. Более мелкие ветви имеют хорошую поддержку. поддержку.