Protein BLAST
Гомологи PriS P.furiosus
Параметры запуска
Последовательность была задана в виде AC из UniProt, поиск осуществлялся в этой же базе данных (UniProtKB/SwissProt). Используемый алгоритм: blastp.
| General Parameters | ||
|---|---|---|
| Max target sequences | E-value threshold | Word size |
| 100 | 0.05 | 6 |
| Scoring Parameters | ||
| Matrix | Gap Costs | Compositional adjustments |
| BLOSUM62 | Existence: 11, Extension: 1 | Conditional compositional score matrix adjustment |
Дополнительно был включён Filter: Low complexity regions.
Результаты
Ознакомиться с выдачей можно по ссылке.
Так как установленный порог E-value довольно низкий (0.05), то в выдаче присутствуют потенциально гомологичные малые субъединицы ДНК-праймаз эукариотического типа других архей. В Jalview было построено выравнивание с алгоритмом Muscle (проект). Последовательности, в целом, можно назвать гомологичными: в них выделяется большое количество консервативных участков и сайтов. Негомологичными являются концевые участки и участки, не учавствующие в функционировании белка.
Выбранные последовательности можно скачать по ссылке.
Гомологи компонента вирусного полипротеина
Для работы был выбран аннотированный полипротеин вируса иммунодефицита человека:
- RecName: Gag-Pol polyprotein
- ID: POL_HV1H2
- AC: P04585; O09777; Q9WJC5
- Organism: Human immunodeficiency virus type 1 group M subtype B (isolate HXB2) (HIV-1)
- Chain: Integrase (1028..1147)
С "вырезанным" фрагментом можно ознакомиться по ссылке.
Параметры запуска BLAST
Полностью повторяют задание 1.
Результаты
Ознакомиться с выдачей можно по ссылке.
Для выравнивания из выдачи BLAST'а были выбраны белки различных вирусов и один белок бактерии:
- P04585: Gag-Pol polyprotein (HIV-1 M:B_HXB2R) - исходно выбранный для сравнения белок
- P12497: Gag-Pol polyprotein (HIV-1 (NEW YORK-5 ISOLATE)) - серотип HIV-1
- P17283: Gag-Pol polyprotein (SIVcpz GAB1) - вирус иммунодефицита обезьян, давший начало HIV [1]
- P19028: Pol polyprotein (Feline immunodeficiency virus (isolate San Diego))
- Q82851: Gag-Pol polyprotein (Jembrana disease virus)
- P32542: Pol polyprotein (Equine infectious anemia virus (clone CL22))
- P16901: Pol polyprotein (Ovine lentivirus (strain SA-OMVV))
- Q82XV8: Ribonuclease H (Nitrosomonas europaea ATCC 19718)
В Jalview с алгоритмом Muscle было построено множественное выравнивание. Последовательности имеют большое количество консервативных участков, поэтому их можно назвать гомологичными, однако RNAse H у N. europaea отличается двумя крупными инделями, что говорит о её сильной дивергенции.
Влияние объема банка на E-value
После установки параметра поиска в пределах таксона Viruses из выдачи пропали все белки бактерий, которые присутствовали в предыдущем варианте. Суммарное количество находок уменьшилось до 77.
Для вычисления доли белков был взят Q82851: Gag-Pol polyprotein (Jembrana disease virus). Необходимые значения из выдачи приведены в таблице ниже.
| Выдача | Score (S) | E-value (Ei) |
|---|---|---|
| 1) All | 79.0 | 4e-17 |
| 2) Viruses | 79.0 | 2e-18 |
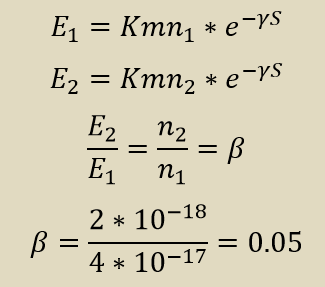
Оценка доли вирусных белков в UniProtKB/SwissProt: 5%
Гомологи случайной последовательности
Несколько случайных последовательностей разной длины были сгенерированы с помощью Python. Они были забластованы на записи с двух баз данных: UniProtKB/Swiss-Prot(sw) и Metagenomic proteins (env_nr) с разным установленными threshold для E-value (0.05 и 10). Дополнительно длина слова поиска была установлена на 6, включена поправка на участки малой сложности.
Результаты выдачи(количество находок с E-value меньше указанного) сведены в таблицу. Если в каком-то случае никаких находок не присутствует, то в таблице стоит -
| Seq | sw + 0.05 | sw + 10 | env_nr + 0.05 | env_nr + 10 |
|---|---|---|---|---|
| YRKSLRFQDIRQKVMEHLPLGLAWS | 3 | 22 | 3 | 25 |
| TATCCVWPCEVHCVMAYTANHQIIHTEDVPRVLPQDVSDK | - | - | - | 1 |
| INYEVMDINTEEEYGAGMKATFIPITVNHSWDVHPHVWDCWFCILISHSG | - | 3 | - | - |
Полученные результаты вполне планомерны: для короткой последовательности находятся совпадения с довольно высоким покрытием (56-92%), однако их E-value говорит об отсутствии гомологии между запросом и выдачей (>1e-2). Тем более, короткие последовательности с большим шансом найдутся в каком-либо белке, а каждое совпадение в выравнивании вносит больший вклад в процент покрытия и Score. Для длинных последовательностей (40-50 аминокислот) все находки имеют E-value больше 1.5, что однозначно говорит об отсутствии гомологии.
Данная работа иллюстрирует "неслучайность" эволюции, которая объясняется целым рядом причин:
- Аминокислотные замены неравновероятны, так что на месте исходной аминокислоты в белке за раунд нуклеотидной замены не может возникнуть любая другая. Конечно, с увеличением времени стохастикой можно было бы получить случайную последовательность, но есть и другие факторы.
- Аминокислоты обладают принципиально разными свойствами (гидрофобность, полярность, заряженность, влияние на вращение полипептидной цепи). Все эти свойства, особенно если аминокислота имеет значение для функционирования белка, не позволяют на месте глицина, например, появиться аргинину. Такой мутантный белок с высокой вероятностью будет негативно влиять на фенотип и элиминируется естественным отбором.
- Аминокислоты образуют "сайты", определяющие функционирование белка. Для осуществления, например, направленного транспорта в матрикс митохондрии нужна целая последовательность определенных аминокислот на N-конце. Аналогично и с активными сайтами: для осуществления ферментативной активности необходимы определенные аминокислоты, находящиеся на определенном расстоянии друг от друга, между которыми находятся определенные остатки, позволяющие свернуться активному центру в нужную конформацию.
- Принцип доменной эволюции белков: в белках выделяются относительно дискретные элементы третичной структуры, которые позволяют ему осуществлять свою функцию. Эти элементы(домены) состоят из определенной последовательности аминокислот и присутствие определенного домена даже в различных по функциям белках свидетельствует об эволюции путем "перестановки" данных блоков.
В целом, это далеко не все причины, но суть остаётся прежней - аминокислотные последовательности белков неслучайны.
REFERENCES
- Sharp P. M., Hahn B. H. Origins of HIV and the AIDS pandemic // Cold Spring Harbor perspectives in medicine. – 2011. – Т. 1. – №. 1. – С. a006841.