Таксоны
Используя таксономический сервис NCBI таблица была дополнена таксонами каждого организма (представлена как табл. 1)
| Название | Мнемоника | Таксон |
|---|---|---|
| Bacillus anthracis | BACAN | Bacilli; Bacillales; Bacillaceae; Bacillus; Bacillus cereus group |
| Bacillus subtilis | BACSU | Bacilli; Bacillales; Bacillaceae; Bacillus |
| Clostridium tetani | CLOTE | Clostridia; Clostridiales; Clostridiaceae; Clostridium |
| Geobacillus kaustophilus | GEOKA | Bacilli; Bacillales; Bacillaceae; Geobacillus |
| Lactobacillus acidophilus | LACAC | Bacilli; Lactobacillales; Lactobacillaceae; Lactobacillus |
| Listeria monocytogenes | LISMO | Bacilli; Bacillales; Listeriaceae; Listeria |
| Staphylococcus epidermidis | STAES | Bacilli; Bacillales; Staphylococcaceae; Staphylococcus |
| Streptococcus pneumoniae | STRPN | Bacilli; Lactobacillales; Streptococcaceae; Streptococcus |
Филогенетическое дерево, где вместо названия бактерии используется её таксономическое положение, представлено на рис. 1.
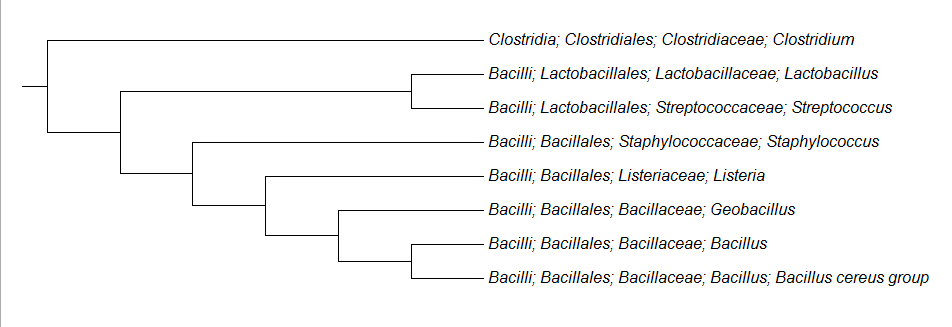
Рис.1 Филогенетическое дерево бактерий из таблицы 1.
Опираясь на полученный рисунок, можно указать 3 ветви, выделяющие таксоны:
- Bacilli - ветвь {Bacilli; *} vs {Clostridia}
- Lactobacillales - ветвь {Bacilli; Lactobacillales; *} vs {Clostridia; *, Bacilli; Bacillales; *}
- Bacillales - ветвь {Bacilli; Bacillales; *} vs {Bacilli; Lactobacillales; *, Clostridia; *}
- Bacillaceae - ветвь {Bacilli; Bacillales; Bacillaceae; *} vs {others}
- Bacillus - ветвь {Bacilli; Bacillales; Bacillaceae; Bacillus} vs {others}
Остальные клады содержат более маленькие таксоны, например:
- Listeriaceae - ветвь {*; Listeria} vs {others}
- Staphylococcaceae - ветвь {*; Staphylococcus} vs {others}
Создание выравнивания
Была выбрана функция белка - Рибосомный белок S2 (RS2), по которому будет проводиться реконструкция. Далее были получены последовательности белков, выполняющих указанную функцию, среди выбранных бактерий (fasta-файл). После проведено выравнивание программой muscle на сервере kodomo - представлено в fasta-файле, на рис. 2 и в проекте JalView.
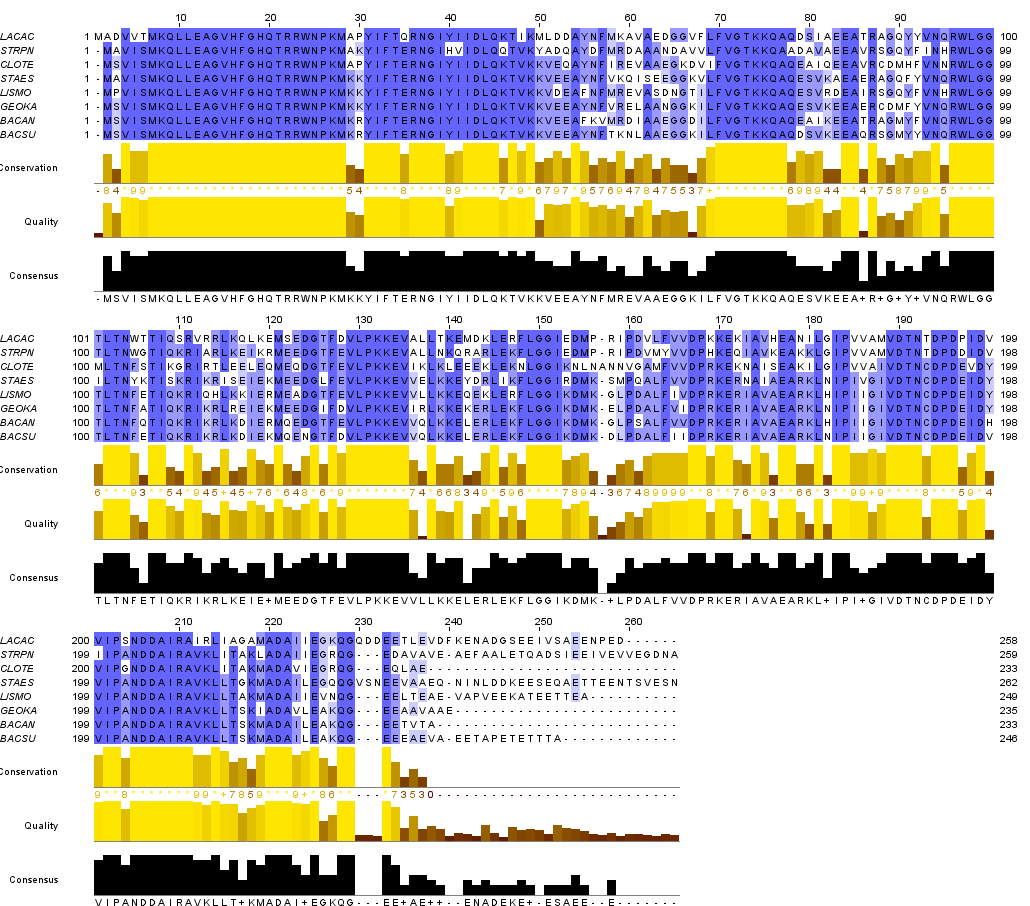
Рис.2 Выравнивание белков семейства RS2, взятых из бактерий из табл. 1
Диагностические позиции
Опираясь на полученные данные, можно выявить следующие диагностические позиции в выравнивании:
- 29K (лизин в позиции 29 выравнивания) свидетельствует о принадлежности к Bacillales
- 140K, 198I (лизин в 140 позиции, изолейцин в 198) — Bacilli
- 122Q (глутамин в позиции 122) — Bacillus
Реконструкция филогенетического дерева
Алгоритмы Average distance и Neigbour Joining
С помощью полученного выравнивания всеми методами, доступными в JalView, была проведена реконструкция дерева. Построенные деревья затем были перерисованы программой MEGA. Результат представлен в таблице 2
| Метод | Average distance | Neigbour Joining |
|---|---|---|
| using % identity | 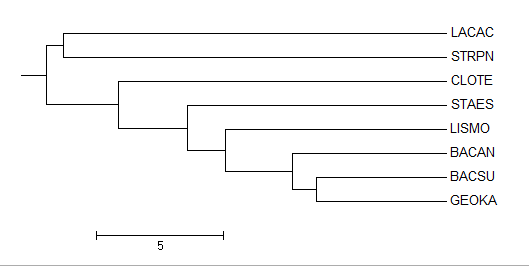
| 
|
| using BLOSUM62 | 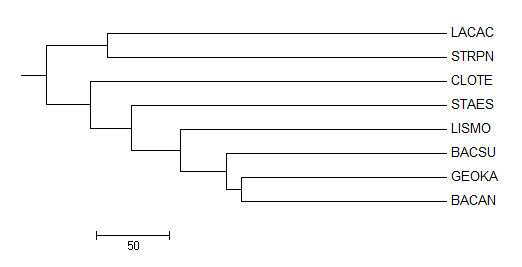
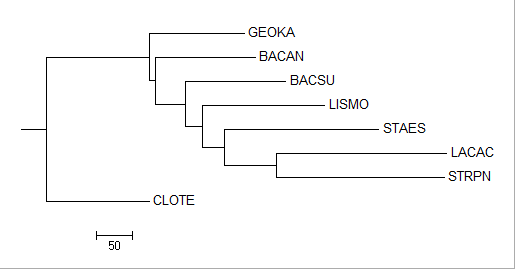
| 
|
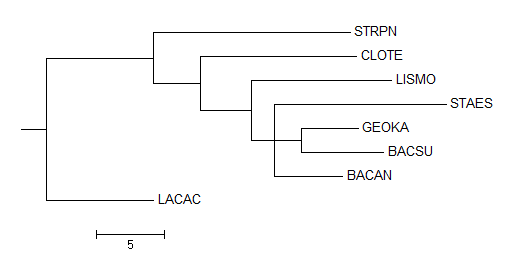
Для удобства работы на рисунке 3 снова приведено правильное дерево:
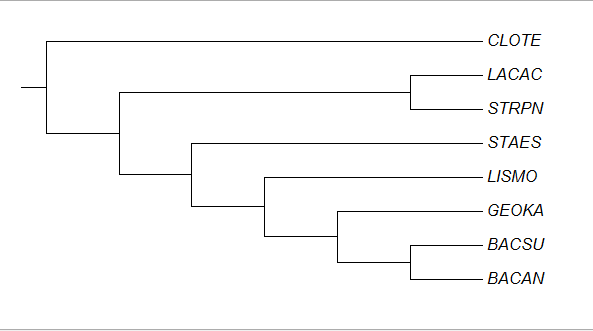
Рис. 3 Правильное дерево в терминах мнемонических названий.
Полученные деревья большей частью повторяют реальное филогенетическое дерево этих бактерий (см. первый раздел). Но есть и отличия:
- Деревья, полученные алгоритмом Average distance почти полностью повторяют настоящую филогению, лишь за разницей, что CLOTE присоединяется к Bacilli, а не выделяется в отдельную ветвь, и наблюдается путаница с выделением таксона Bacillus (GEOKA оказывается ближе то к BACSU, то к BACAN, а должен быть к ним одинаково близок)
- Деревья, построенные с помощью алгоритма Neigbour Joining, куда хуже отражают действительность. Дерево, для которого использовалась BLOSUM62 отражает всё с точностью до наоборт. Второе же, во-первых, не бинарно; во-вторых, оно не выделяет таксон Lactobacillales; в-третьих, здесь опять же неверно определяется таксон Bacillus.
Более строгое сравнение деревьев представлено в таблице 3.
| Ветвь | Правильное дерево | Neigbour Joining using BLOSUM62 | Average distance using BLOSUM62 | Neigbour Joining using % identity | Average distance using % identity | Maximum Parsimony |
|---|---|---|---|---|---|---|
| {LACAC, STRPN} vs {others} | + | + | + | + | + | + |
| {CLOTE, LACAC, STRPN} vs {others} | + | - | + | + | + | + |
| {LISMO, GEOKA, BACSU, BACAN} vs {others} | + | - | + | - | + | - |
| {GEOKA, BACSU, BACAN} vs {others} | + | - | + | - | + | - |
| {BACSU, BACAN} vs {others} | + | - | - | - | - | - |
| {GEOKA, BACAN} vs {others} | - | - | + | - | - | - |
| {BACSU, GEOKA} vs {others} | - | - | - | + | + | + |
| {STAES, GEOKA, BACSU, BACAN} vs {others} | - | - | - | + | - | + |
| {STAES, LISMO, LACAC, STRPN} vs {others} | - | + | - | - | - | - |
| {STAES, LACAC, STRPN} vs {others} | - | + | - | - | - | - |
| {CLOTE, GEOKA} vs {others} | - | + | - | - | - | - |
| {CLOTE, GEOKA, BACAN} vs {others} | - | + | - | - | - | - |
| {GEOKA, BACSU, STAES} vs {others} | - | - | - | - | - | + |
| Верных ветвей | 5 | 1 | 4 | 2 | 4 | 2 |
| Ошибок | 0 | 4 | 1 | 2 | 1 | 3 |
Полученное выравнивание импортировано в программу MEGA, с помощью которой затем построено филогенетическое дерево методом Maximum Parsimony. После оно было укоренено в ветвь, разделяющую классы. Результаты представлены в таблице 4.
| До укоренения | После укоренения |
|---|---|
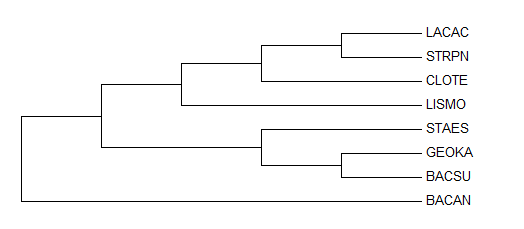
| 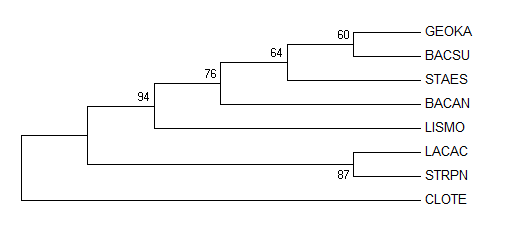
|
Полученное после укоренения дерево имеет схожие отличия, что и предыдущие реконструкции (см. табл. 3):
- Правильно выделяется таксон Lactobacillales
- Опять произошла путаница с Bacillaceae
- STAES оказался близок к Bacillaceae, в то время как должен был разойтись с ними ещё раньше, чем LISMO
Обсуждение
Анализ количества мутаций только в белке RS2 не даёт правильного филогенетического дерева, поэтому необходимо проводить анализ по нескольким белкам. Однако построенные деревья дают хоть и приблизительное, но представление о филогении выбранных организмов.
Согласно таблице 3 лучше всего себя показали деревья, построенные методом Average distance.