| FBB MSU site | Main page | About me | Terms |
Resequencing
Searching for polymorphisms in humans
Part I
Task 0
I created a directory /nfs/srv/databases/ngs/potanina.darya. This directory contains reads and chromosome files and all prodused during making the tasks ones.
Task 1 and 2
Firstly, I conducted quality control of reads by FastQC installed on kodomo. The result you can see here: chr9_1_fastqc.html.
| $ fastqc chr9_1.fastq |
Secondly, I sorted reads. It was made by Trimmomatic installed on kodomo with parameters TRAILING:20 (to cut off end less then 20 nucleotides quality) and MINLEN:50 (to leave only more then 50 nucleotides length reads).
| $ java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr9_1.fastq chr9_1_cleaned.fastq TRAILING:20 MINLEN:50 |
Output (from 10701 reads 10536 were survived and 165 were dropped):
| TrimmomaticSE: Started with arguments: -phred33 chr9_1.fastq chr9_1_cleaned.fastq TRAILING:20 MINLEN:50 Automatically using 8 threads Input Reads: 10701 Surviving: 10536 (98,46%) Dropped: 165 (1,54%) TrimmomaticSE: Completed successfully |
Then I made quality control again. The result here: chr9_1_cleaned_fastqc.html.
| $ fastqc chr9_1_cleaned.fastq |
As we can see quality of reads was increased after trimming of ends: average value increased and lenght of whiskers decreased which means reducing the number of drop-down values (see fig. 1,2).
| Per base sequence quality | |
| Figure 1. Quality of reads before cleaning. | Figure 2. Quality of reads after cleaning. |
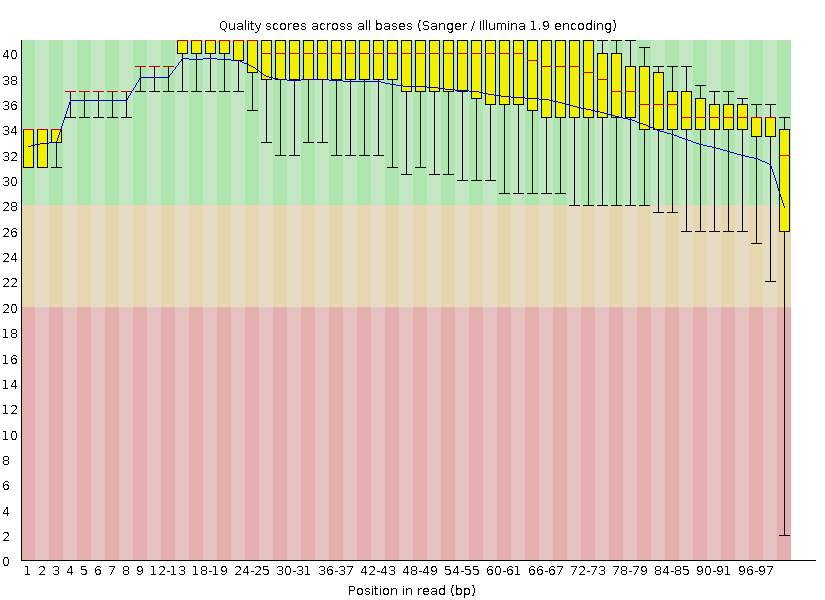 | 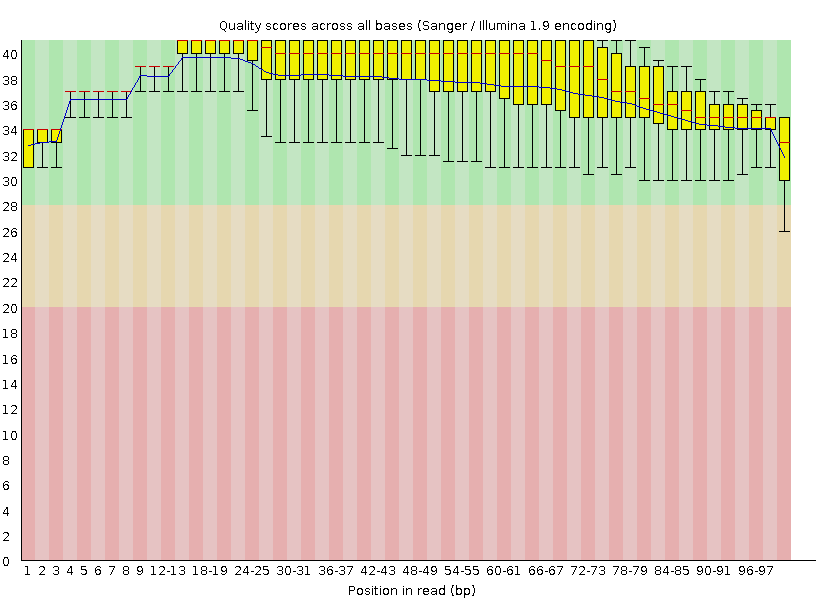 |
Part II
Task 3
Cleaned reads were scanned by HISAT2. Firstly the reference sequence was indexed. The protocol of the program execution: hisat2.txt.
| $ export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 $ hisat2-build chr9.fasta chr9_indexed |
As a result of program execution 8 files were created they are named chr9_indexed.*.ht2 (*=1,2,...8).
Then I run hisat2 with parametres --no-spliced-alignment and --no-softclip. In the end, there was made an alignment of the reference sequence and purified reads in SAM format.
| $ hisat2 -x chr9_indexed -U chr9_1_cleaned.fastq --no-spliced-alignment --no-softclip -S chr9_alignment.sam |
Parametres:
--no-softclip - Disallow soft-clipping
--no-spliced-alignment - Disable spliced alignment (gaps are forbidden)
-x - The basename of the index for the reference genome.
-U - input file with reads
-S - output file in SAM format
Output:
| 10536 reads; of these: 10536 (100.00%) were unpaired; of these: 74 (0.70%) aligned 0 times 10459 (99.27%) aligned exactly 1 time 3 (0.03%) aligned >1 times 99.30% overall alignment rate |
Task 4
Analisys of the alignment was made by samtools. Firstly, alignment was converted from SAM to BAM format.
| $ samtools view chr9_alignment.sam -b -o chr9_alignment.bam |
Secondly, I sort the alignment of readings with the reference and the coordinate of the beginning reference reading (*_temp.txt is a temporary file created by the program).
| $ samtools sort chr9_alignment.bam -T chr9_alignment_temp.txt -o chr9_alignment_sort.bam |
Thirdly, sequence in the resulting file was indexed.
| $ samtools index chr9_alignment_sort.bam |
Part III
Task 5
I created file with polynorphisms
| $ samtools mpileup -uf chr9.fasta chr9_alignment_sort.bam -o chr9_snp.bcf |
By bcftools was created a file with a list of differences between reference sequense and reads.
| $ bcftools call -cv chr9_snp.bcf -o chr9_snp.vcf |
In VCF file I found a few SNPs (see table 5.1). In total there are 5 indels and 106 snp.
Table 5.1.
| # | Type of polimorphism | Coordinates | Reference | Read | Depth | Quality |
| 1 | Replasing | 5093614 | G | T | 3 | 4.12848 |
| 2 | Indel | 5122854 | agg | ag | 11 | 95.4652 |
| 3 | Replasing | 136135238 | T | C | 15 | 222 |
Task 6
For SNP annotation I deleted all indels from chr9_snp.vcf and saved it as chr9_snp_6.vcf. Then I converted the new one from VCF to AVINPUT format by annovar.
| $ perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 chr9_snp_6.vcf -o chr9.avinput |
Output (106 SNPs: 73 transitions* and 33 transversions** and 0 indels/substitutions):
|
NOTICE: Finished reading 138 lines from VCF file NOTICE: A total of 106 locus in VCF file passed QC threshold, representing 106 SNP (73 transitions and 33 transversions) and 0 indels/substitutions NOTICE: Finished writing 106 SNP genotypes (73 transitions and 33 transversions) and 0 indels/substitutions for 1 sample |
| $ perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr9_snp_rs -build hg19 -dbtype snp138 chr9.avinput /nfs/srv/databases/annovar/humandb/ |
Output (amount of rs 51):
| NOTICE: Variants matching filtering criteria are written to chr9_snp_rs.hg19_snp138_dropped, other variants are written to chr9_snp_rs.hg19_snp138_filtered NOTICE: Processing next batch with 106 unique variants in 106 input lines NOTICE: Database index loaded. Total number of bins is 2894320 and the number of bins to be scanned is 51 NOTICE: Scanning filter database /nfs/srv/databases/annovar/humandb/hg19_snp138.txt...Done |
Output files:
|
chr9_snp_rs.hg19_snp138_dropped chr9_snp_rs.hg19_snp138_filtered chr9_snp_rs.log |
Then these 106 SNPs were annotate using differnt databases.
1. Refgene
| $ perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr9_refgene -build hg19 chr9.avinput /nfs/srv/databases/annovar/humandb/ |
Output:
|
NOTICE: The --geneanno operation is set to ON by default NOTICE: Reading gene annotation from /nfs/srv/databases/annovar/humandb/hg19_refGene.txt ... Done with 50914 transcripts (including 11516 without coding sequence annotation) for 26271 unique genes NOTICE: Reading FASTA sequences from /nfs/srv/databases/annovar/humandb/hg19_refGeneMrna.fa ... Done with 5 sequences WARNING: A total of 345 sequences will be ignored due to lack of correct ORF annotation NOTICE: Finished gene-based annotation on 106 genetic variants in chr9.avinput NOTICE: Output files were written to chr9_refgene.variant_function, chr9_refgene.exonic_variant_function |
Output files:
|
chr9_refgene.exonic_variant_function chr9_refgene.log chr9_refgene.variant_function |
Table 6.1. Locations of SNPs.
| Area | Amount of SNPs | Comment |
| intronic | 78 | variant overlaps an intron |
| exonic | 15 | variant overlaps a coding |
| UTR3 | 8 | variant overlaps a 3' untranslated region |
| intergenic | 3 | variant is in intergenic region |
| downstream | 2 | variant overlaps 1-kb region downtream of transcription end site (use -neargene to change this) |
Table 6.2. SNPs in exons.*
| Coordinates | Gene | exon:aa replacemetn:nucleotide replacement | Ref | Read | Synonymy | Quality | DP | Genotype |
| 5081780 | JAK2:NM_004972 | exon19:c.G2490A:p.L830L | G | A | synonymous | 221.999 | 50 | hom |
| 5126443 | JAK2:NM_004972 | exon24:c.T3288A:p.D1096E | T | A | nonsynonymous | 207.009 | 38 | het |
| 6253571 | IL33:NM_001199641 | exon3:c.C111T:p.Y37Y,IL33:NM_033439:exon6:c.C489T: p.Y163Y,IL33:NM_001199640:exon5:c.C363T:p.Y121Y | T | C | synonymous | 225.009 | 54 | het |
*There are 12 SNPs in unknow areas (due to various errors in the gene structure definition in the database file).
2. dbsnp
| $ perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype snp138 -out chr9_dbsnp -build hg19 chr9.avinput /nfs/srv/databases/annovar/humandb/ |
Output:
|
NOTICE: Variants matching filtering criteria are written to chr9_dbsnp.hg19_snp138_dropped, other variants are written to chr9_dbsnp.hg19_snp138_filtered NOTICE: Processing next batch with 106 unique variants in 106 input lines NOTICE: Database index loaded. Total number of bins is 2894320 and the number of bins to be scanned is 51 NOTICE: Scanning filter database /nfs/srv/databases/annovar/humandb/hg19_snp138.txt...Done |
Output files:
|
chr9_dbsnp.hg19_snp138_dropped chr9_dbsnp.hg19_snp138_filtered chr9_dbsnp.log |
3. 1000 genomes
| $ perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -out chr_1000genomes -buildver hg19 chr9.avinput /nfs/srv/databases/annovar/humandb/ |
Output:
|
NOTICE: Variants matching filtering criteria are written to chr_1000genomes.hg19_ALL.sites.2014_10_dropped, other variants are written to chr_1000genomes.hg19_ALL.sites.2014_10_filtered NOTICE: Processing next batch with 106 unique variants in 106 input lines NOTICE: Database index loaded. Total number of bins is 2824642 and the number of bins to be scanned is 51 NOTICE: Scanning filter database /nfs/srv/databases/annovar/humandb/hg19_ALL.sites.2014_10.txt...Done |
Output files:
|
chr_1000genomes.hg19_ALL.sites.2014_10_dropped chr_1000genomes.hg19_ALL.sites.2014_10_filtered chr_1000genomes.log |
4. GWAS
| $ perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -dbtype gwasCatalog -out chr9_gwas -buildver hg19 chr9.avinput /nfs/srv/databases/annovar/humandb/ |
Output:
|
NOTICE: Reading annotation database /nfs/srv/databases/annovar/humandb/hg19_gwasCatalog.txt ... Done with 18027 regions NOTICE: Finished region-based annotation on 106 genetic variants in chr9.avinput NOTICE: Output file is written to chr9_gwas.hg19_gwasCatalog |
Output files:
|
chr9_gwas.hg19_gwasCatalog chr9_gwas.log |
Table 6.3. The responsibility of the SNPs for various diseases.
| Disease | Coordinate | Ref | Read | Genotype | Quality | DP |
| Crohn's disease | 4985879 | A | G | het | 225.009 | 30 |
| Endometriosis | 6253571 | C | T | het | 225.009 | 54 |
| Tumor biomarkers, Coagulation factor levels | 136131188 | C | T | het | 13.2188 | 10 |
| mean corpuscular hemoglobin concentration | 136131322 | G | T | het | 180.009 | 24 |
| End-stage coagulation | 136131415 | C | T | het | 134.008 | 21 |
| Malaria | 136132754 | C | A | het | 123.008 | 33 |
| D-dimer levels, Venous thromboembolism, Activated partial thromboplastin time | 136137065 | A | G | hom | 42.7648 | 2 |
| Coagulation factor levels | 136137106 | G | A | hom | 9.52546 | 1 |
5. Clinvar
| $ perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype clinvar_20150629 -out chr9_clinvar -buildver hg19 chr9.avinput /nfs/srv/databases/annovar/humandb/ |
Output:
|
NOTICE: the --dbtype clinvar_20150629 is assumed to be in generic ANNOVAR database format NOTICE: Variants matching filtering criteria are written to chr9_clinvar.hg19_clinvar_20150629_dropped, other variants are written to chr9_clinvar.hg19_clinvar_20150629_filtered NOTICE: Processing next batch with 106 unique variants in 106 input lines NOTICE: Database index loaded. Total number of bins is 49139 and the number of bins to be scanned is 7 NOTICE: Scanning filter database /nfs/srv/databases/annovar/humandb/hg19_clinvar_20150629.txt...Done |
Output files:
|
chr9_clinvar.hg19_clinvar_20150629_dropped chr9_clinvar.hg19_clinvar_20150629_filtered chr9_clinvar.log |
Table 6.4. Clinical interpretation of the SNPs.*
| CLNDBN | Coordinates | Ref | Read | Genotype | Quality | DP |
| ABO_BLOOD_GROUP_SYSTEM | 136131315 | C | G | het | 204.009 | 24 |
| ABO_BLOOD_GROUP_SYSTEM | 136131651 | G | A | het | 4.12848 | 3 |
*There is not_specified SNP in 5126443 position.
The summary result you can see in summary table (click the link, pls).
| Term 3 |
| ← Block 2 | Pr 12 → |
© Darya Potanina, 2017