| FBB MSU site | Main page | About me | Terms |
Analysis of the transcriptome
Bedtools (↓)
I created a subdirectory /nfs/srv/databases/ngs/potanina.darya/pr12. This directory contains two samples of RNA sequencing reads derived from human's chromosome 9 and all prodused during making the tasks files.
All comands you can find here: script.sh.
Task 1
I conducted quality control of reads by FastQC installed on kodomo.
| $ fastqc chr9.1.fastq $ fastqc chr9.2.fastq |
The result you can see here:
Output files:
|
chr9.1_fastqc.html - the first replica chr9.2_fastqc.html - the second replica |
| Per base sequence quality | |
| Figure 1. First replica. | Figure 2. Second replica. |
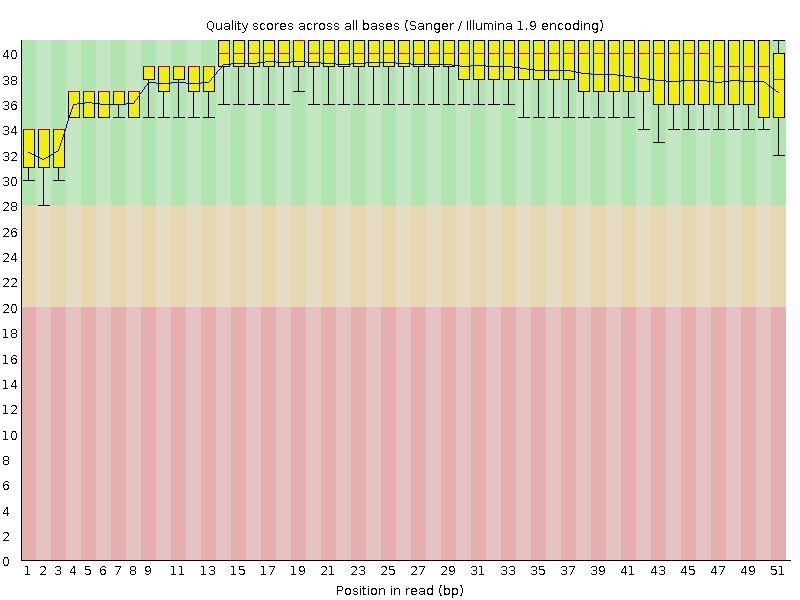 | 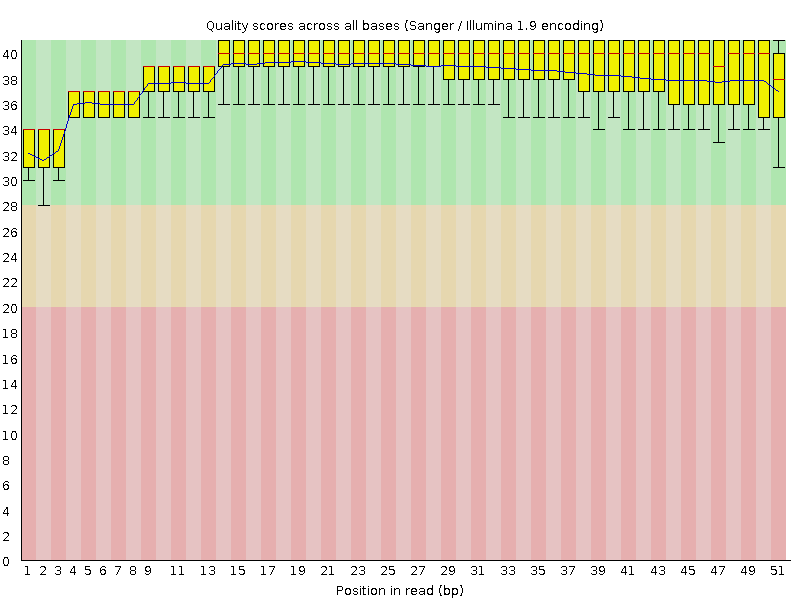 |
| We can observe high quality of reads (are entirely in the green area). | |
| Per Illumina tile sequence quality | |
| Figure 3. First replica. | Figure 4. Second replica. |
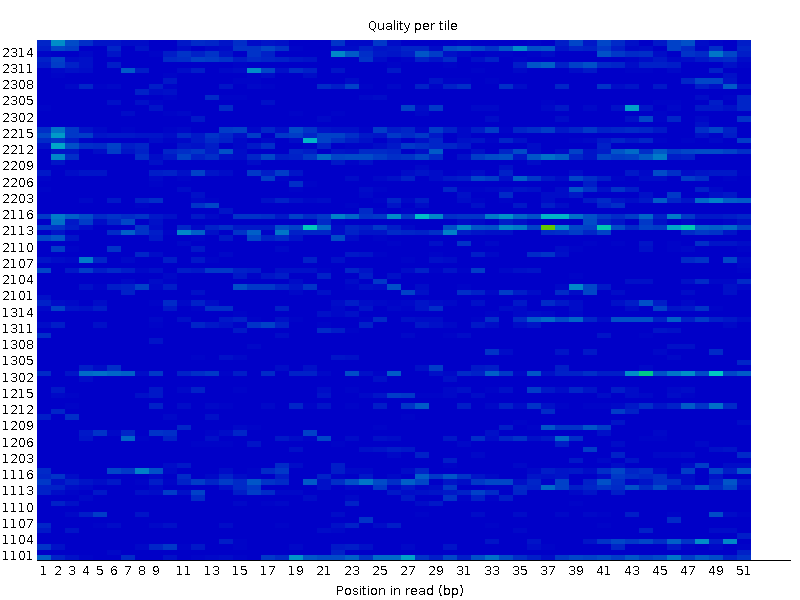 | 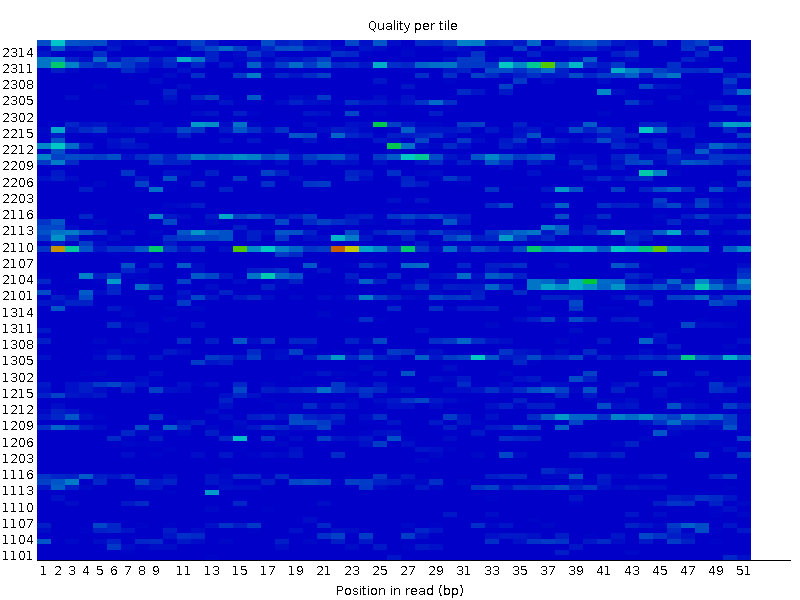 |
| The quality of reads in the cells colored blue is the highest and lower the cells colored yellow and red. As we can see the most of reads are high quality, but there are a few poorly read ones and even one very poor in the second replica. In general, the quality of the second replica is higher than the first. | |
| Per base sequence content | |
| Figure 5. First replica. | Figure 6. Second replica. |
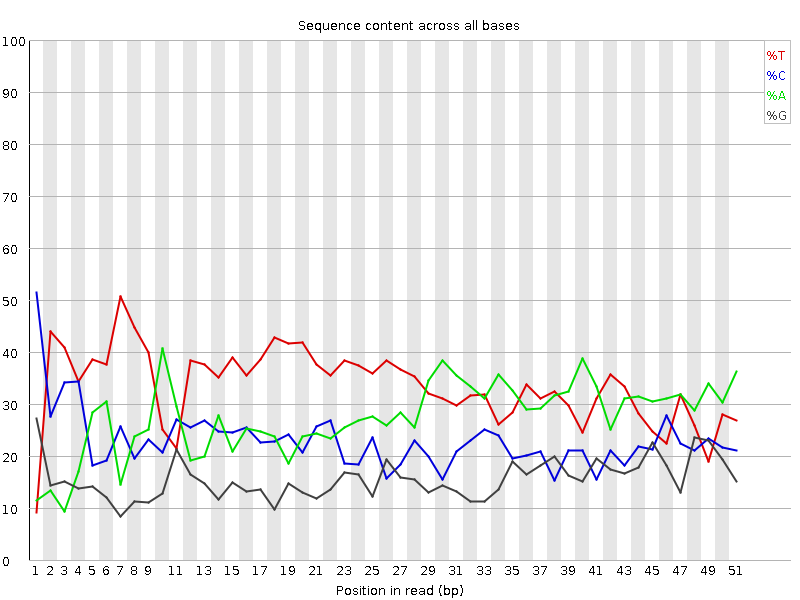 | 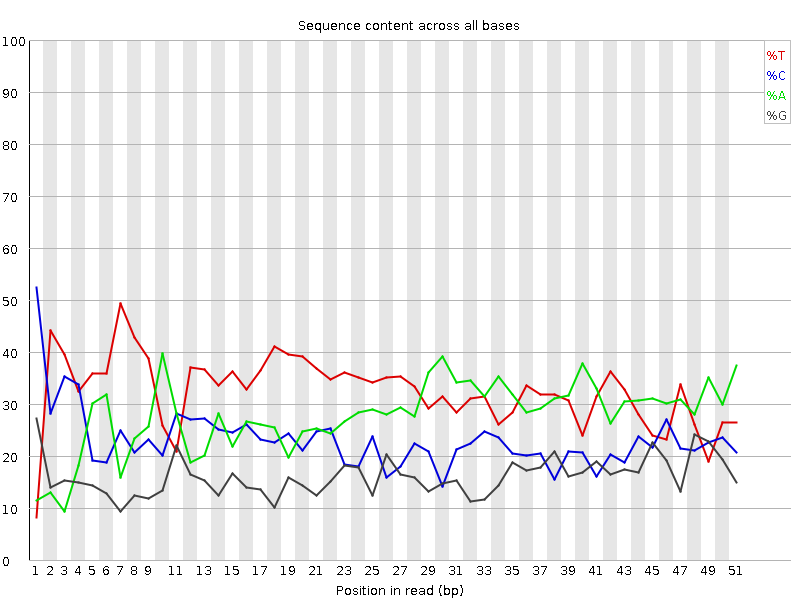 |
| The average amount of A and T is greater than the average number of C and G in each position along the reads. We can also notice that the number of T in the positions along the first half of the reads is increased. | |
| Per sequence GC content | |
| Figure 7. First replica. | Figure 8. Second replica. |
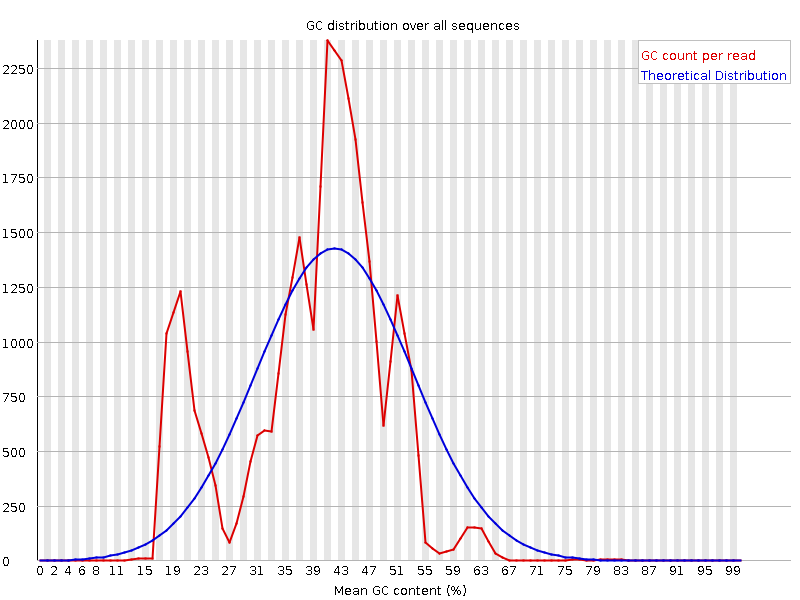 | 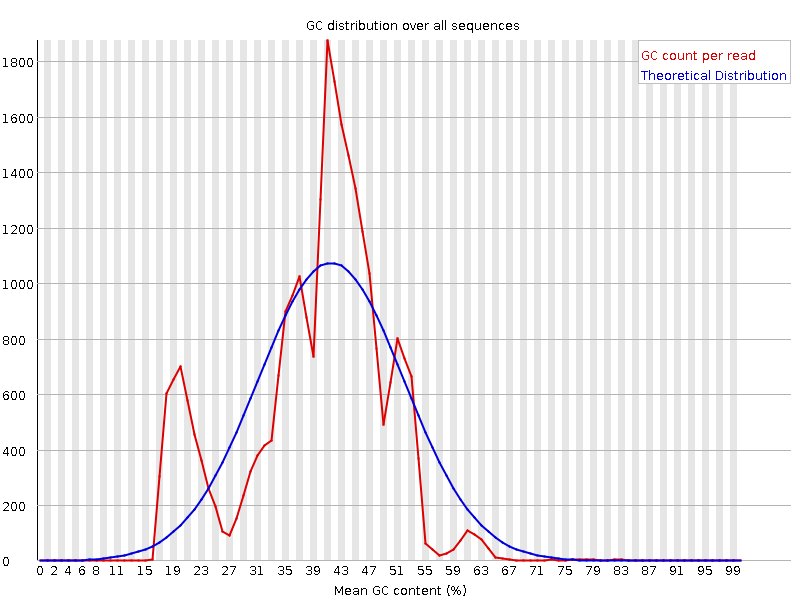 |
| The amount of GC content is much higher than expected in 40ths and 20ths positions, but it is lower then expected in 25ths and 55ths positions. | |
Task 2
Reads were mapped on chromosome 9 by HISAT2. I run hisat2 without parameter --no-spliced-alignment because gaps are not forbidden for transcriptome reads (the indexed reference sequense was taken from the previous work). Then there was made an alignment of the reference sequence and readings in SAM format.
| $ export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 |
| $ hisat2 -x chr9_indexed -U chr9.1.fastq --no-softclip -S chr9.1.sam $ hisat2 -x chr9_indexed -U chr9.2.fastq --no-softclip -S chr9.2.sam |
Output (here you can see the number of unaligned reads, reads aligned once and reads aligned more than once):
|
1st replica: 19976 reads; of these: 19976 (100.00%) were unpaired; of these: 115 (0.58%) aligned 0 times 19680 (98.52%) aligned exactly 1 time 181 (0.91%) aligned >1 times 99.42% overall alignment rate 2nd replica: 14020 reads; of these: 14020 (100.00%) were unpaired; of these: 83 (0.59%) aligned 0 times 13811 (98.51%) aligned exactly 1 time 126 (0.90%) aligned >1 times 99.41% overall alignment rate |
Output files:
|
chr9.1.sam chr9.2.sam |
Task 3.
Analisys of the alignment was made by samtools. Firstly, alignment was converted from SAM to BAM format.
| $ samtools view chr9.1.sam -b -o chr9.1.bam $ samtools view chr9.2.sam -b -o chr9.2.bam |
Output files:
|
chr9.1.bam chr9.2.bam |
Secondly, I sort the alignment of readings with the reference and the coordinate of the beginning reference reading (*_temp.txt is a temporary file created by the program).
| $ samtools sort chr9.1.bam -T chr9.1_temp.txt -o chr9.1_sorted.bam $ samtools sort chr9.2.bam -T chr9.2_temp.txt -o chr9.2_sorted.bam |
Output files:
|
chr9.1_sorted.bam chr9.2_sorted.bam |
Thirdly, sequence in the resulting file was indexed.
| $ samtools index chr9.1_sorted.bam $ samtools index chr9.2_sorted.bam |
Output files:
|
chr9.1_sorted.bam.bai chr9.2_sorted.bam.bai |
Task 4
I conducted reads' counting by bedtools. Firstly BAM files were converted to BED format.
| $ bedtools bamtobed -i chr9.1_sorted.bam > chr9.1.bed $ bedtools bamtobed -i chr9.2_sorted.bam > chr9.2.bed |
Output files:
|
chr9.1.bed chr9.2.bed |
Then compared RNA sequencing reads with the reference sequence of human genome h19 assembly (/nfs/srv/samba/public/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed). This program allows one to screen for overlaps between two sets of genomic features. It was run with the following parametres:
-a - the first BED file
-b - the second BED file
-u - reports the fact at least one overlap was found in B.
grep - to extract chr9 only from the whole genome
| $ export PATH=${PATH}:/P/y14/term3/block4/SNP/bedtools2/bin $ bedtools intersect -a gencode.genes.bed -b chr9.1.bed -c | grep 'chr9' > chr9.1_intersect.bed $ bedtools intersect -a gencode.genes.bed -b chr9.2.bed -c | grep 'chr9' > chr9.2_intersect.bed |
Output:
|
***** WARNING: File /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed has inconsistent naming convention for record: GL877870.2 2348 2545 pseudogene CICP10 ***** WARNING: File /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed has inconsistent naming convention for record: GL877870.2 2348 2545 pseudogene CICP10 ***** WARNING: File /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed has inconsistent naming convention for record: GL877870.2 2348 2545 pseudogene CICP10 ***** WARNING: File /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed has inconsistent naming convention for record: GL877870.2 2348 2545 pseudogene CICP10 |
Output files:
|
chr9.1_intersect.bed chr9.2_intersect.bed |
There are gene types: [antisense, IG_C_pseudogene, IG_V_pseudogene, lincRNA, miRNA, misc_RNA, polymorphic_pseudogene, processed_transcript, protein_coding, rRNA, sense_intronic, sense_overlapping, snoRNA, snRNA, TR_V_gene, TR_V_pseudogene, pseudogene] - in the final files. The list of a few genes covered by reads you can see in table 1.
Table 1
| Gene | Start | End | Amount of reads in the 1st replica | Amount of reads in the 2nd replica | Gene function |
| CBWD1 | 121041 | 188979 | 235 | 235 | COBW domain-containing protein 1 |
| DOCK8 | 214854 | 465259 | 438 | 438 | Dedicator of cytokinesis 8 - a protein involved in intracellular signalling networks. |
| SMARCA2 | 2015342 | 2193624 | 326 | 326 | Probable global transcription activator |
| SPATA6L | 4553386 | 4666674 | 248 | 248 | Spermatogenesis associated 6 like |
| MPDZ | 13105703 | 13279589 | 652 | 652 | Multiple PDZ domain protein |
Some additional files (columns: amount of reads covered the region, gene type, gene name):
The list of genes covered by reads for 1st replica: chr9.1_genes_list.csv.txt
The list of genes covered by reads for 2nd replica: chr9.2_genes_list.csv.txt
The list of protein coding genes covered by reads for 1st replica: chr9.1_prot_cod.csv.txt
The list of protein coding genes covered by reads for 1st replica: chr9.2_prot_cod.csv.txt
I noted that two final BED files differ very little. Therefore "csv.txt" files are the same. I can't explain why it is so. A list of different lines you can find here. The result is that almost all genes are covered by the same number of reads.
Bedtools
Task 1
According the task I need to extract reads in FASTQ format from the file contain alignment.
| $ bedtools bamtofastq -i chr9.1_sorted.bam -fq chr9.1.fq |
Output files:
| chr9.1.fq |
Task 2
According the task I need to receive FASTA file of one of the genes covered by reads from the whole chromosome. I saved coordinates of the gene in BED file CBWD1.bed. Then I used getfasta function of Bedtools.
| $ bedtools getfasta -fi chr9.fasta -bed CBWD1.bed > CBWD1.fasta |
Output files:
| CBWD1.fasta |
Task 3
According the task I need to break the chromosome into fragments of 1 million nucleotides. Informatoin about chromosome length I checked here and save it in chr9.genome. Total tenth of the chromosome = 141213431 nucleotodes. Amount of received fragments is 142.
| $ bedtools makewindows -g chr9.genome -w 1000000 > chr9_1mln_fragments.bed |
Output files:
| chr9_1mln_fragments.bed |
Task 4
According the task I need to merge reads into clusters using BED file contains alignment from the previous task (pr 12). Parameter -d means the maximum distance between reads to merge them in the same clusters.
| $ bedtools cluster -i chr9.1.bed -d 10000 > clusters1.bed |
Output files (there are only 1 cluster even if use -d 100000):
| clusters1.bed |
Task 5
According the task I need to make 1000 random fragments of 200 nucleotides from the chromosome. Informatoin about chromosome lenth I checked here and save it in chr9.genome.
| $ bedtools random -g chr9.genome -n 1000 -l 200 > chr9_random.bed |
Output files:
| chr9_random.bed |
Task 7
According the task I need to recieve coordinates of a gene covered by reads expanded to 1000 nucleotides in right and left. I saved coordinates of the gene in BED file CBWD1.bed. Then I used slop function of Bedtools with parameter -b = 1000 (increase the BED/GFF/VCF entry -b base pairs in each direction).
| $ bedtools slop -i CBWD1.bed -g chr9.genome -b 1000 |
Output:
| chr9 120041 189979 |
Task 8
According the task I need to recieve coordinates of a gene covered by reads shifted 500 nucleotides closer to the start of the chromosome. I saved coordinates of the gene in BED file CBWD1.bed. Then I used slop function of Bedtools with parameters -l = 500 (the number of base pairs to subtract from the start coordinate) and -r = 500 (the number of base pairs to add to the end coordinate).
| $ bedtools slop -i CBWD1.bed -g chr9.genome -l 500 -r -500 |
Output files:
| chr9 120541 188479 |
Task 9
According the task I need to obtain non-overlapping fragments which correspond to the regions covered by 'my' genes. I used BED file contains alignment from the previous task (pr 12) and this file (human genome assembly h19): /nfs/srv/samba/public/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed.
| $ bedtools annotate -i chr9.1.bed -files gencode.genes.bed -both > annotation1.bed |
Output files:
| annotation1.bed |
| Term 3 |
| ← Pr 11 | Pr 14 → |
© Darya Potanina, 2017