








Формат Fastq используется для записи результатов секвенирования. Он содержит в себе последовательности ридов и качество прочтения каждой позиции. Программа FastQC обрабатывает файл fastq и выдает различные графики и таблицы, показывающие качество секвенирования.
Я работала с файлом с чтениями генома резуховидки Ath_tae_CTTGTA_L003_R1_005.fastq.gz. Отчет программы был сохранен в html форме: fastqc_before_trimming.html.
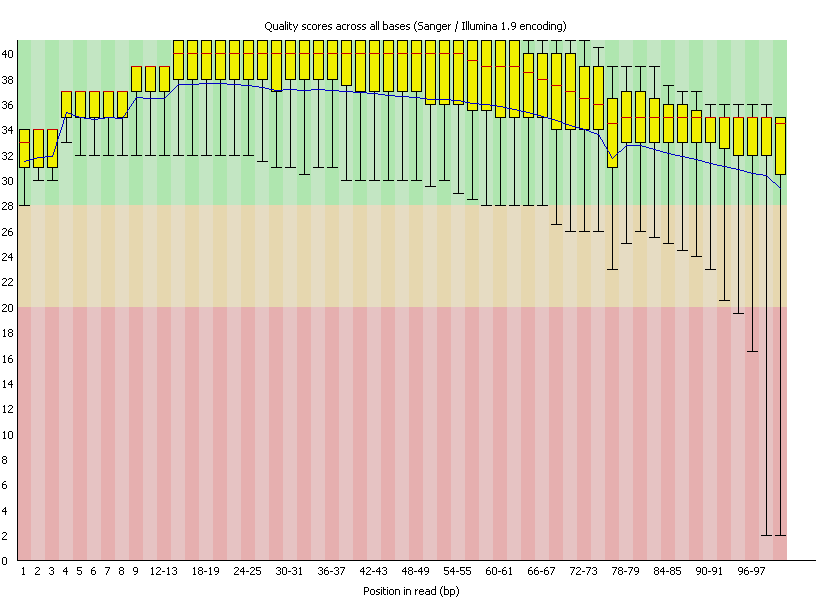
Результаты секвенирования можно очистить программой Trimmomatic. В командной строке подаем Trimmomatic phred33, т.к. в FastQC видим, что сиквенс сделан Illumina 1.9, а после 1.8 качество делается в phred33 [x]. ILLUMINACLIP - параметр, указывающий файл с праймерами, использовавшимися в процессе секвенирования - чтобы убрать случайно прочтенные праймеры из файла. Параметр TRAILING убирает с конца ридов нуклеотиды с качеством меньше заданного. MINLENGTH убирает все риды длиной меньше данной. Использованная команда выглядела так:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 Ath_tae_CTTGTA_L003_R1_005.fastq.gz Trimmomatic_cleaned.fastq ILLUMINACLIP:adapters.fa:2:7:7 TRAILING:20 MINLEN:50
Очищенный файл был снова подан FastQC. Результаты: Trimmomatic_cleaned_fastqc.html
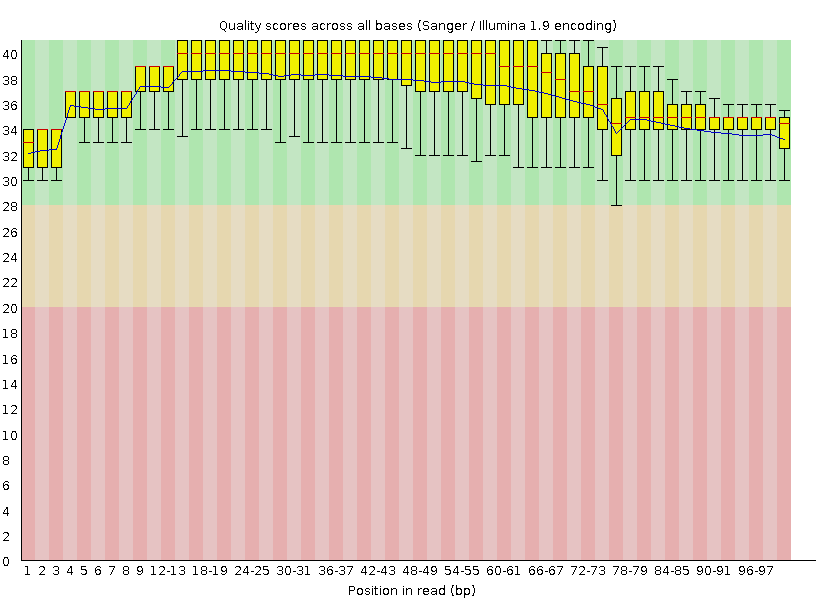
Видно, что разброс по качеству уменьшился, особенно заметно в конце ридов, потому что мы убирали нуклеотиды с плохим качеством с концов ридов. Ридов стало 3841341 вместо 4000000. Per base sequence content, однако, почти не изменился. Распределение длин было равномерным, но после очистки стало много коротких ридов, поскольку мы убирали нуклеотиды с концов.