Семестры
Сайт ФББ МГУ
Kodomo Wiki
NCBI
RanHummer personal web-site
BLAST
Для выполнения заданий данного практикума была взята аминокислотная последовательность белка Mutator MutT из Mesorhizobium opportunistum WSM2075.
Задание 1. Найти сходные последовательности в базе данных UniProtKB/SwissProt.
В данном задании было необходимо найти последовательности из базы данных UniProtKB/SwissProt, сходные с исходной последовательностью белка.
Для этого на сайте NCBI я выбрал программу BLAST, которая ищет схожие с искомой последовательности, затем выбрал protein blast, так как нужно найти сходные аминокислотные последовательности белков. Загрузил последовательность моего белка в fasta-формате, затем в окне Database выбрал базу данных UniProtKB/SwissProt, в поле Algorithms выбрал blastp (protein-protein BLAST), так как мы ведем поиск по последовательности белка. Параметры алгоритма были оставлены по умолчанию (штрафы за гэпы, ожидаемый порог по E-value, матрица и др.). Ссылка на запрос
Для того, чтобы определить, какое число находок какой группе организмов принадлежит, нужно перейти по ссылке Formatting options, там в поле Organisms выбрать нужную группу, затем кнопка Reformat.
В результате такого запроса было получено 330 находок (Sbjct): 274 белков из организма бактерий, 2 из архей, 42 из эукариот.
В Таблице 1 для лучшей, худшей находок и находки из середины списка были сравнены важнейшие показатели выравнивания:
- Длина выравнивания
- bit score - характеристика, показывающая, насколько выравнивание "хорошее" (т. е. насколько сильно совпадает с Query), и нормированная на особенности матрицы. Чем выше bit score, тем лучше выравнивание.
- Процент идентичных остатков
- Процент сходных остатков
- E-value - число находок с таким же или лучшим Score в случайном банке (чем оно меньше, тем лучше выравнивание)
Лучшая находка, безусловно, первая, худшая - последняя, и еще одна случайно выбрана из середины списка.
| Название белка | Организм | Длина выравнивания | Bit score | Процент идентичных колонок, % | Процент сходных колонок, % | E-value |
|---|---|---|---|---|---|---|
| CTP pyrophosphohydrolase | Escherichia coli K-12 | 133 | 94.7 | 41 | 48 | 2e-19 |
| Uncharacterized Nudix hydrolase orf19 | Rhodococcus erythropolis | 95 | 40.1 | 34 | 38 | 0.014 |
| Uncharacterized Nudix hydrolase NudL | Klebsiella pneumoniae subsp. pneumoniae MGH 78578 | 47 | 30.9 | 43 | 48 | 9.6 |
Ниже приведены сами выравнивания выбранных последовательностей с исходной (Рис.1-3).
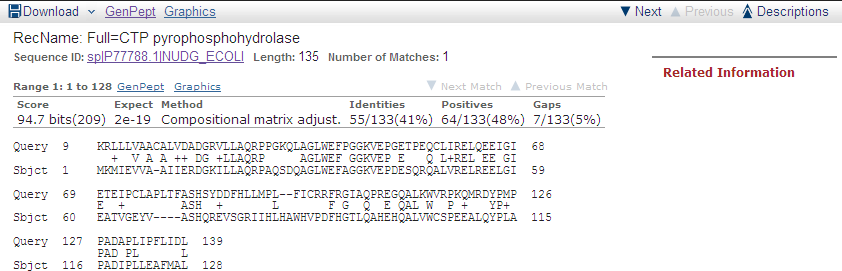
Рис. 1. Выравнивание последовательности белка CTP pyrophosphohydrolase из организма Escherichia coli K-12 с исходной.
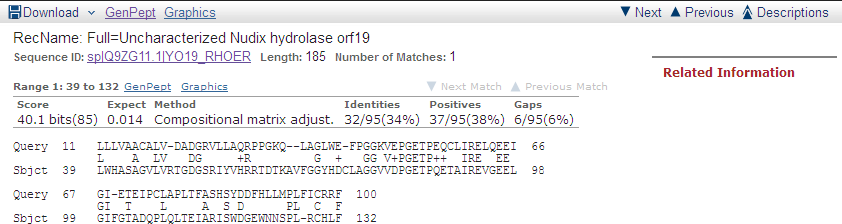
Рис. 2. Выравнивание последовательности белка Uncharacterized Nudix hydrolase orf19 из организма Rhodococcus erythropolis с исходной.
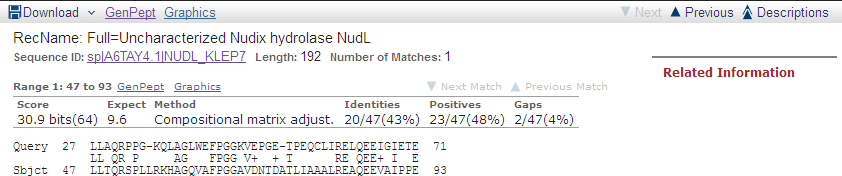
Рис. 3. Выравнивание последовательности белка Uncharacterized Nudix hydrolase NudL из организма Klebsiella pneumoniae subsp. pneumoniae MGH 78578 с исходной.
Из всех полученных находок гомологами исходной последовательности можно считать 35. По условному критерию последовательность можно считать гомологичной, если E-value < 1e-3 и не менее 70% запроса вошло в полученное выравнивание (Query cover). Для того, чтобы найти находки-гомологи, я задал максимальный E-value 0.001 в Formatting options, отсортировал находки по убыванию Query cover и вручную отобрал нужные.
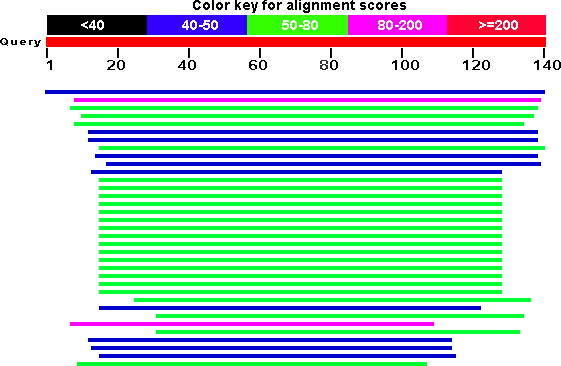
Рис. 4. Графическое представление находок-гомологов.
Однако вышеупомянутый критерий не всегда является истинным. Может быть так, что происходят довольно обширные индели при дивергенции организмов, поэтому значение Query cover будет ниже 70%, но гомологичность нельзя будет отрицать (особенно если организмы принадлежат к родственным группам). Поэтому с высокой вероятностью можно говорить о гомологичности последовательностей, если это один и тот же белок у родственных организмов, практически невзирая на значение Query cover.
Задание 2. Найти сходные последовательности среди белков из какой-нибудь таксономической группы.
В этом задании я искал схожие с исходной последовательности в таксоне Escherichia coli. Для этого я вернулся на страницу с параметрами запроса и в поле Organisms указал Escherichia coli (Условия поиска). Там среди находок я выбрал последовательность CTP pyrophosphohydrolase, которая была также найдена в первоначальном запуске (для первоначального результата с помощью окна Formatting options я отобрал находки, относящиеся к Escherichia coli (Organism: Escherichia coli)). О том, что это та же самая находка, свитедельствуют одинаковые название, Sequence ID (Accession): P77788.1, длина найденной последовательности (Length: 135).
У этих находок полностью совпадают выравнивание и Score, отличается только E-value: для первого запуска 2e-19, для второго запуска 3e-21. Такое изменение объяснимо: E-value показывает, насколько случайна полученная находка, и так как второй банк был меньше, то данная находка менее случайна.
Задание 3. Для одной из найденных последовательностей сохраните карту локального сходства.
Для последовательности CTP pyrophosphohydrolase из организма Escherichia coli K-12 (идентификатор в UniProtKB - P77788.1) было выполнено выравнивание (Условия поиска) и получена локальная карта сходства (Рис. 5):
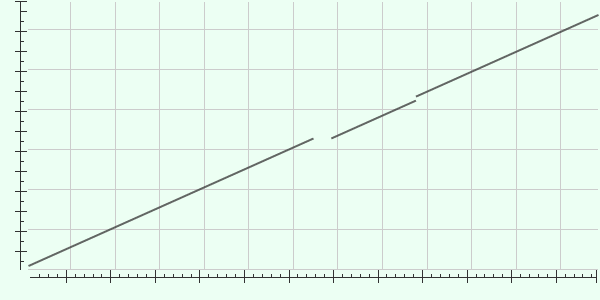
Рис. 5. Локальная карта сходства последовательности белка CTP pyrophosphohydrolase из организма Escherichia coli K-12 с исходной.
На Рис. 5 видны некоторые особенности полученного выравнивания (Рис. 1):
- Последовательности совпадают почти на всем протяжении со сдвигом после середины (Query cover 91%).
- Первая половина выравнивания очень хороша (линия на рисунке сплошная) - содержит много консервативных и сходных позиций и содержит только 1 гэп.
- Сразу после середины выравнивания присутствуют 2 гэпа средней протяженности, соответствующие инделям (пробелы на графике).
Задание 4. Используйте BLAST для поиска в своей базе данных.
Для выполнения данного задания я создал свою базу данных, полученную из выравнивания последовательностей из файла align_03.fasta, предварительно удалив все гэпы (файл my_align.fasta). Для этого на сервере kodomo я запустил команду makeblastdb -dbtype prot -in my_align.fasta -out my_align - так был получен банк, содержащий 6 последовательностей. Затем я совершил поиск последовательностей, схожих с моим белком (файл my.fasta) в полученной базе данных (команда blastp -db my_align -query my.fasta). Всего программа признала значимыми 5 находок: участки последовательностей MACCJ (2 находки), CELLD, LEUCK, BREBN, ETHHY. Для лучшей находки (MACCJ 1, Рис. 6) перечислены основные характеристики: длина выравнивания, % идентичных и сходных остатков, Bit score и E-value:
- Длина выравнивания: 85
- Ident: 29%
- Positives: 44%
- Bit score: 17.7
- E-value: 0.3
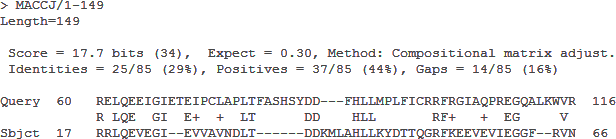
Рис. 6. Выравнивание последовательности белка Mutator MutT из Mesorhizobium opportunistum WSM2075 (идентификатор YP_004610222) с последовательностью MACCJ из нового банка.
О параметрах выравнивания по новой базе данных можно сказать то, что матрица и штрафы за гэпы совпадают с предыдущими запусками, но появляется такой параметр, как Neighboring words threshold со значением 11, а также есть параметр, относящийся к окну выдачи результатов (Window for multiple hits: 40).
Проанализировав в целом числовые данные и выраванивания, можно заключить, что длина выравниваний меньше, чем в предыдущих запусках программы (для большинства - значительно меньше), Bit score довольно небольшой, процент идентичных и сходных колонок примерно такой же (если длина выравнивания больше 20 остатков) или намного больше, чем в предыдущих выравниваниях, так как полученные выравнивания слишком маленькие. Что касается E-value, то его значение слишком большое для нового банка (при предыдущих запусках при уменьшении банка уменьшался и E-value), даже самое маленькое значение из имеющихся больше 0.1. Так как E-value большой, а Score маленький, то можно судить об отсутствии гомологии, так как полученные совпадения с высокой вероятностью случайны.