I
Мною был выбран домен, который называется CHASE (PF03924). Это внеклеточный домен, который есть в трансмембранных белках-рецепторов как у эукариот, так и у бактерий. Его полное название — Cyclases/Histidine kinases Associated Sensory Extracellular. Было показано, что данный домен отвечает за связывание низкомолекулярных лигандов (таких как цитокинин-подобные производные аденина) или небольших пептидов и может опосредовать проведение сигнала в связанном с ним рецепторе.
У выбранного семейства имеется 392 варианта архитектуры, представленных 1724 последовательностями из 890 видов. Первые семь архитектур по количеству последовательностей показаны на рисунке ниже.

Исходя из рисунка, видно, что доменные архитектуры бывают совершенно разные: так, в белках с доменом CHASE могут присутствовать и другие домены: EAL, PAS_3, GGDEF и проч. Для дальнейшей работы были выбраны две следующие доменные архитектуры: [CHASE, GGDEF] и [CHASE, HisKA, HATPase_c, Response_reg].
| I | II | |
|---|---|---|
| Схема | ||
| Число последовательностей | 156 | 263 |
| Характеристика сторонних доменов |
|
|
При помощи следующей команды была получена информация об архитектуре всех последовательностей, содержащих CHASE-домен:
python swisspfam-to-xls.py -w -i /srv/databases/pfam/swisspfam.gz -p PF03924 -o architectures.xls
После этого из этой выдачи с использованием Uniprot / Retrieve была получена информация с таксономией:
python uniprot-to-taxonomy.py -i uniprot.txt -o taxa.xlsИнформация, полученная в ходе работы, была скомбинирована в общую сводную таблицу (лист называется pivot). В этой таблице указаны последовательности, их доменные архитектуры, таксономии и длины CHASE-домена. Для дальнейшей работы я выбрал кладу Proteobacteria с двумя её подгруппами — α-протеобактериями и β-протеобактериями (в таблице это обозначено префиксами A и B).
Протеобактерии — очень интересная группа бактерий. Так, известно, что α-протеобактериями дали начало всем митохондриям эукариот (в результате симбиоза, который А. Марков назвал «великим», археи и этой самой бактерии). Это доподлинно известно: так, если построить дерево различных бактерий и митохондрий эукариот (например, по гены 16S рРНК), то будет видна монофилия альфа-протеобактерий и митохондрий. Бета-протеобактерии гораздо менее интересны в плане симбиозов, но тоже представяют достаточно крупную группу с разнообразными бактерийми — как гетеротрофами, так и фото- и хемоавтотрофами.
Для выполнения следующего задания были выбраны определённые последовательности, которые представлены в той же Excel-таблице на листе chosen. После было построено множественное выравнивание (его проект — в нём три окна с различными выравниваниями: до и после «чистки»). Из общего выравнивания были удалены плохо выравнивающиеся и короткие последовательности. Изображение выравнивания представлено ниже.
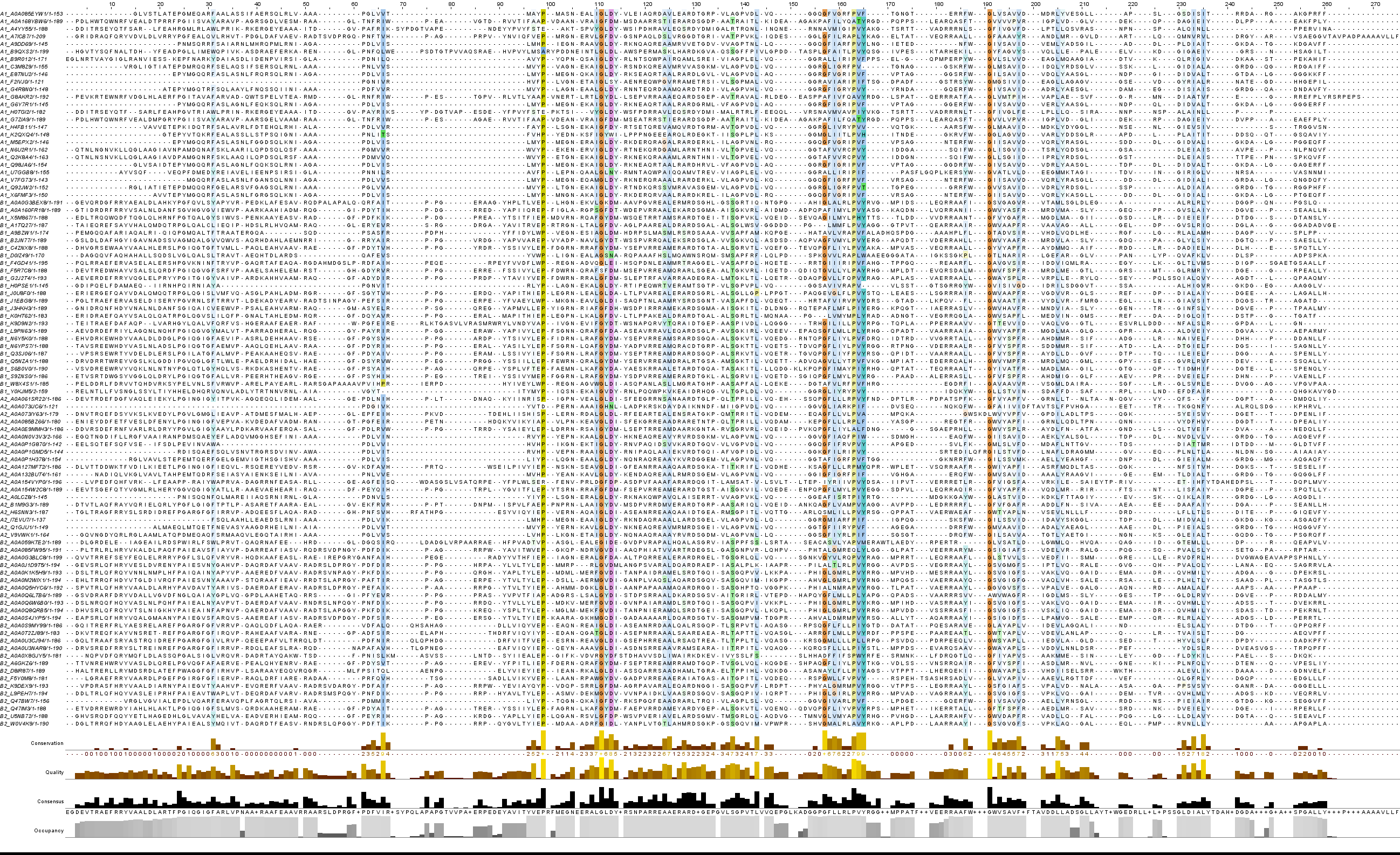
II
По полученному выравниванию было построено филогенетическое дерево (использовался метод Neighbor-Joining). Его изображение представлено ниже. Красными ветвями обозначены α-протеобактерии, а синими — β-протеобактерии.
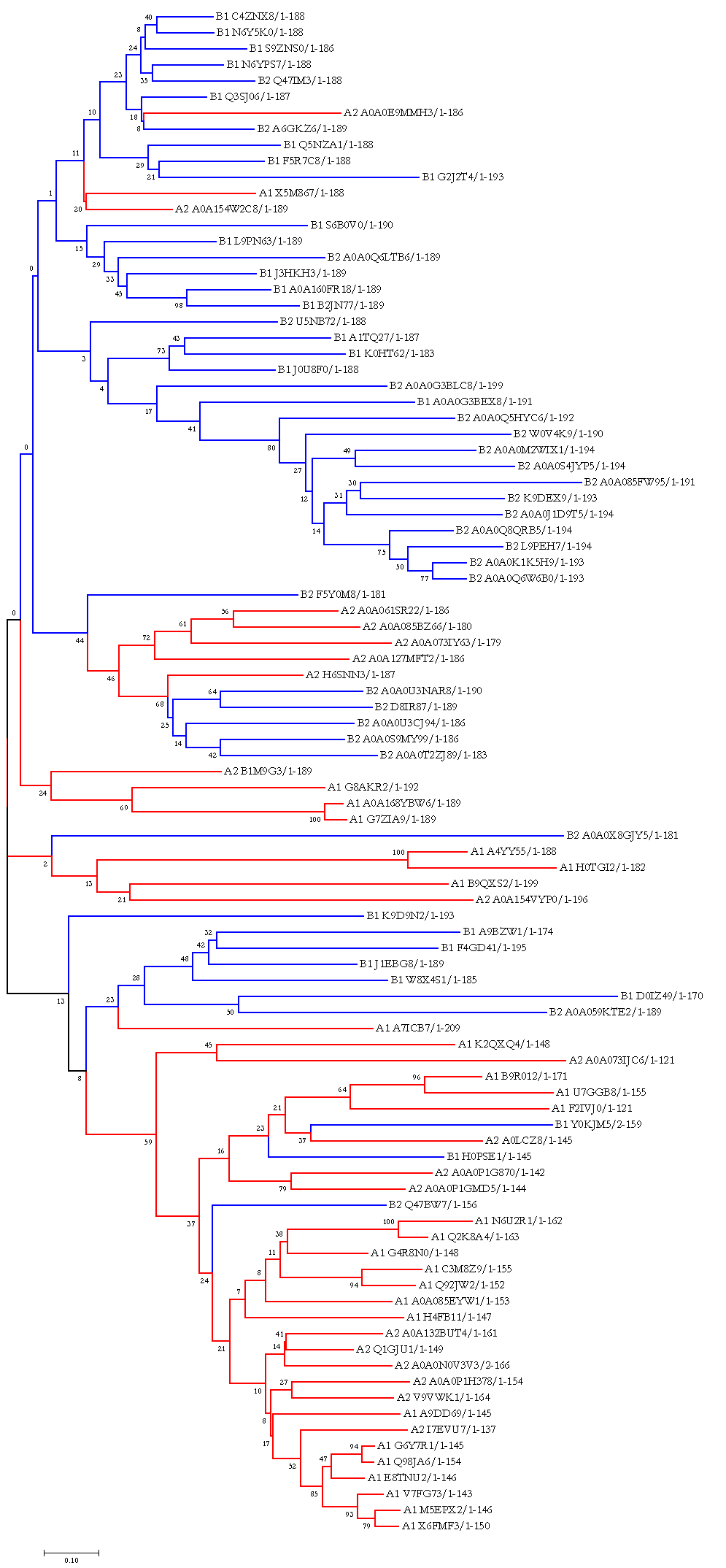
В целом этому дереву, скорее, можно доверять, чем нельзя: видно достаточно много нетривиальных ветвей (причём порой достаточно крупных), состоящих из исключительно представителей какой-то определённой группы. Однако есть и менее однозначеные клады: их существование можно объяснить двумя факторами. Во-первых, в целом дерево получается с достаточно маленькой бутстреп-поддержкой, особенно в подобных спорных моментах, поэтому только опираясь на него говорить о каких-либо высоковероятных родственных отношениях нельзя. Во-вторых, для бактерий характерен горизонтальный перенос генов (ГПГ), благодаря которому генетический материал может распространяться не только «вертикально», то есть от особи к её потомкам, но и между далекородственными видами.
Если же говорить об эволюции именно доменов, то какой-то чёткой кластеризации по разным архитектурам не заметно, по данному дереву сложно сказать о небольшом количестве каких-то дискретных преобразований, которые бы привели к различным архитектурам. Возможно, что сходство обусловлено конвергенцией, но говорить об этом тоже сложно. Для дополнительного анализа необходимо построение деревьев с последовательностями других доменов из представленных доменных архитектур, что не входит в рамки данного практикума.