| Команда | Функция | Результат |
|---|---|---|
| fastqc chr4.1.fastq | Анализ качества необработанного секвенирования | Отчет и zip-архив с его составляющими |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr4.1.fastq chr4trimmed1.fastq TRAILING:20 MINLEN:48 | Обрезка с конца чтений нуклеотидов с качеством меньше 20 и последующее отбрасывание чтений короче 48 | Файл chr4trimmed1.fastq |
| fastqc chr4trimmed1.fastq | Анализ качества обрезанного секвенирования | Отчет и архив с соответсвующими изображениями |
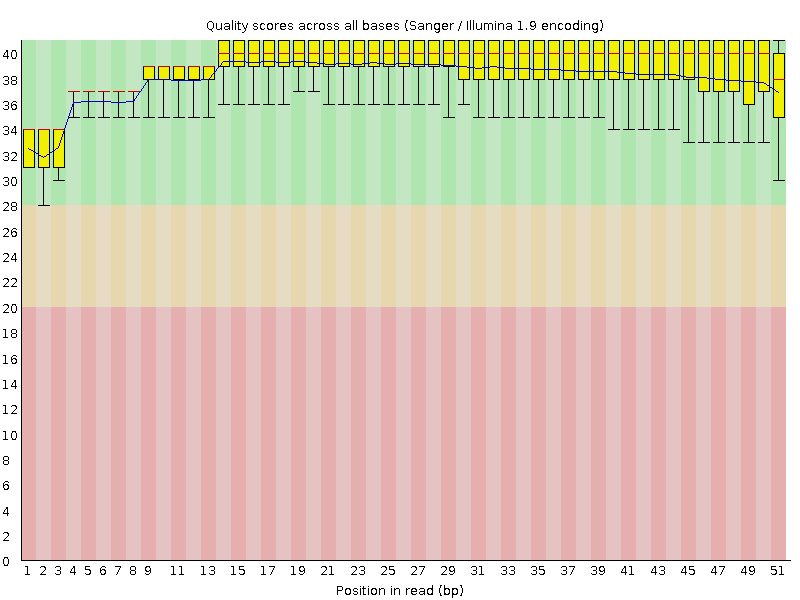
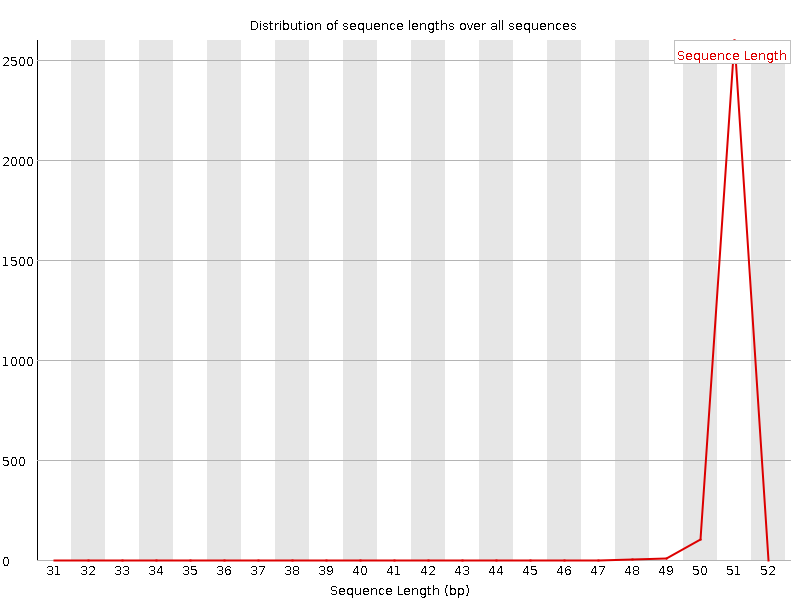
На подготовительном этапе был произведен сбор статистики по начальному материалу, очистка концев и отсев коротких чтений, а потом сбор новой статистики. (аналогично предыдущему практикуму)
| Параметр | Исходное | Подготовленное |
|---|---|---|
| Качество | | |
| Число чтений | 2735 | 2724 |
| Длина чтений | | |
Комментарий: был проведен отсев чтений короче 48 чтобы отсеить выбивающиеся более, чем на два стандартных отклонения. Обрез по низкому качеству концов практически не поменял картину.
| Команда | Функция | Результат |
|---|---|---|
| PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 | Добавление пакета программ в path | Программы становятся дотупны для вызова через командную строку. |
| hisat2-build chr4ref.fasta chr4 | Индексирование референсной последовательности | Несколько файлов расширения ht2 |
| hisat2 -x chr4 -U chr4trimmed1.fastq --no-softclip > alignment.sam | Создание выравнивания референсной последовательности и прочтений | Выравнивание в формате sam |
| samtools view alignment.sam -bo alignment.bam | Перевод выравнивания в бинарный формат | Выравнивание в формате bam |
| samtools sort alignment.bam -T out_sort.txt -o alignment_sorted.bam | Сортировка бинарного выравнивания | Отсортированное выравнивание |
| samtools index alignment_sorted.bam | Индексирование бинарного выравнивания | Проиндексированное отсортированное выравнивание |
В результате выравнивания 2647 прочтения были мапированы на референс однократно, пять более одного раза, а 72 не попали вообще. Программа hisat2 вызывалась без аттрибута --no-splice-alignment так как человек - эукариотический организм и обладает механизмами постранскрипционной обработки РНК, включающие сплайсинг. Таким образом, соседние в РНК фрагменты последовательности могут быть разделены интронами в геноме.
Для подсчета чтений использовался пакет Bedtools. Файл с разметкой был перенесен в рабочую директорию. По итогам пересечения, результирующий файл был достаточно маленький для подсчетавручную.
| Команда | Функция | Результат |
|---|---|---|
| PATH=${PATH}:/P/y14/term3/block4/SNP/bedtools2/bin | Добавление пакета программ в path | Программы становятся дотупны для вызова через командную строку. |
| bedtools bamtobed -i alignment_sort.bam > step1.bed | Конвертация файла в формат BED | Файл c переведенными в bed формат последствиями выравнивания |
| bedtools intersect -a marked.bet -b step1.bed -u > step2.bed | Файл с геномами пересекается с файлом с прочтениями, приэтом репортируются только имеющие более одного пересечения строки | Файл c найденными генами |
| Кординаты | Ген | Функция | Направление цепи | Кол-во чтений |
|---|---|---|---|---|
| 10069713:10074643 | RP11-448G15.3 | Кодирует фермент - глицерол-3-фосфатацетилтрансферазу, участвующий в регуляции липидного обмена. | + | 3 |
| 10075963:10118573 | WDR1 | Кодирует белок, состоящий из 9 доменов, осуществляющих белок-белковые взаимодействия. Участвует в разборке актиновых филаментов в клетке. | + | 226 |
| 10080235:10080316 | MIR3138 | МикроРНК 3138 | + | 3 |
| 10117380:10117508 | RNA5SP155 | Рибосомальная 5S РНК | + | 3 |
Итоги работы с программами пакета bedttols изложены на отдельной странице.