Хромосома № 8
| cp Human/rnaseq_reads/chr8.2.fastq sofyagdk26/12_rna_chr8_2.fastq | Копирование файла с транскриптомными ридами |
|---|---|
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 12_rna_chr8_2.fastq 12_chr8_trimmed.fastq TRAILING:20 MINLEN:50 TrimmomaticSE: Started with arguments: -phred33 12_rna_chr8_2.fastq 12_chr8_trimmed.fastq TRAILING:20 MINLEN:50 | Триммирование чтений |
| fastqc 12_chr8_trimmed.fastq | Запуск анализа FastQ - потом в html файл за анализом |
| hisat2 -x indexed -U 12_chr8_trimmed.fastq -S 12_chr8_altoref.sam --no-softclip | Был убран параметр --no-spliced-alignment, тк мы картируем транскриптомыне данные - а при прицессинге мРНК происходит сплайсинг - поэтому мы и выравнивем с инделями, зная, что сплайсинг есть и будет |
| samtools view -b 12_chr8_altoref.sam -o 12_chr8_altoref.bam | Перевод в бинарный файл |
| samtools sort 12_chr8_altoref.bam 12_chr8_altoref_sorted | Сортировка |
| samtools index 12_chr8_altoref_sorted.bam | индексация |
| htseq-count -i gene_id -s no -m union -f bam 12_chr8_altoref_sorted.bam /nfs/srv/databases/ngs/Human/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf -o 12_c8_union.sam | Подсчет чтений. -i - нужен для задания индекса -s - специфично ли исследование к считываемой цепочке (или необходимо анализировать оба направления считывания - тогда пишется "no") -f - формат входного файла - bam/sam -m - способ подсчета: union; intersection-strict - только если чтение легло на ген целиком; intersection-nonempty - если чтение имеет общую последовательность с геном |
Изначально было 17507 чтений.
Результат работы Trimmomatic:
Input Reads: 17507 Surviving: 17398 (99,38%) Dropped: 109 (0,62%)До обрезки
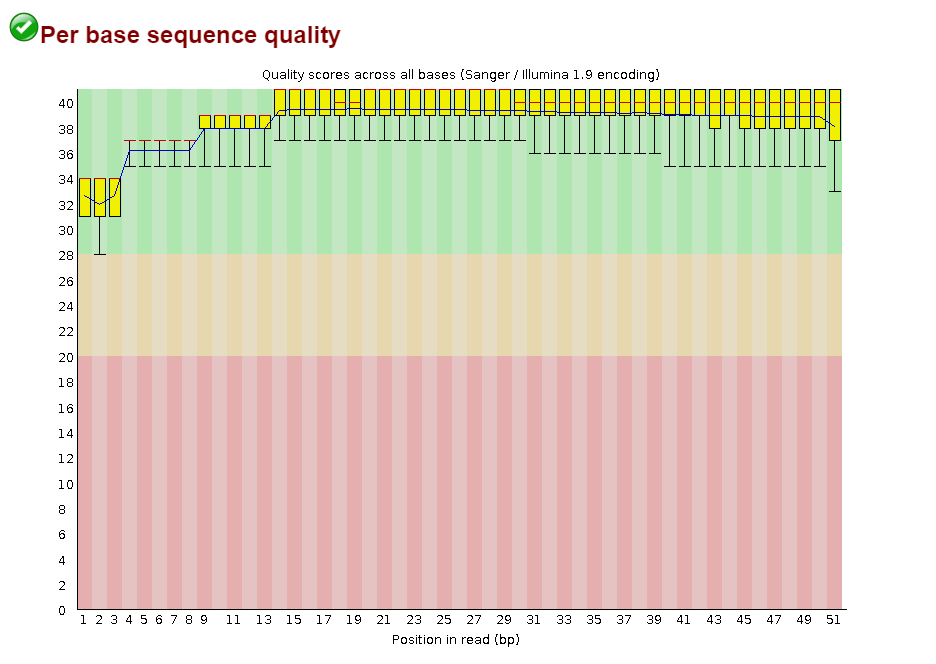
Откартировано 98.01% чтений
Вывод программы hisat2:
17398 reads; of these:
17398 (100.00%) were unpaired; of these:
346 (1.99%) aligned 0 times
17052 (98.01%) aligned exactly 1 time
0 (0.00%) aligned >1 times
98.01% overall alignment rate
Вывод: Кажется, это очень хорошее качество картирования!
Я сделала запуск с тремя разными параметрами. Файлы выдачи обсчитала командой
"sort 12_c8_union.sam | uniq -c"
Выдачи union и nonempty совпали. А вот strict, ожидаемо, оставил больше последовательностей
неподсчитанными (примерно на треть).
| -m union |
__no_feature 1243 __ambiguous 0 __too_low_aQual 0 __not_aligned 346 __alignment_not_unique 0 |
204 XF:Z:ENSG00000104738.12
15605 XF:Z:ENSG00000253729.3
1243 XF:Z:__no_feature
346 XF:Z:__not_aligned
|
| -m intersection-strict |
__no_feature 1676 __ambiguous 0 __too_low_aQual 0 __not_aligned 346 __alignment_not_unique 0 |
188 XF:Z:ENSG00000104738.12
15188 XF:Z:ENSG00000253729.3
1676 XF:Z:__no_feature
346 XF:Z:__not_aligned
|
| -m intersection-nonempty |
__no_feature 1243 __ambiguous 0 __too_low_aQual 0 __not_aligned 346 __alignment_not_unique 0 |
204 XF:Z:ENSG00000104738.12
15605 XF:Z:ENSG00000253729.3
1243 XF:Z:__no_feature
346 XF:Z:__not_aligned
|