|
Home page Term 1 Term 2 Term 3 About me Faculty website |
Transcriptome reads mappingTHERE IS A TABLE WITH ALL THE COMMANDS USED AT THE END OF THE WEBPAGERead quality analysisAccording to the output of the FastQC program, the given single-end reads have a satisfactory quality, despite the fails in "Per base sequence content" which may be attributed to primers, "Per sequence GC content", which can be explained by the unrepresentativeness of the whole genome region, "Sequence duplication levels", "Overrepresented sequences", and also a warning in the "Kmer content" section of the FastQC report, which can all be ascribed to the fact that it was RNA sequencing.FastQC report The main merit of the given set of reads is their initial quality: 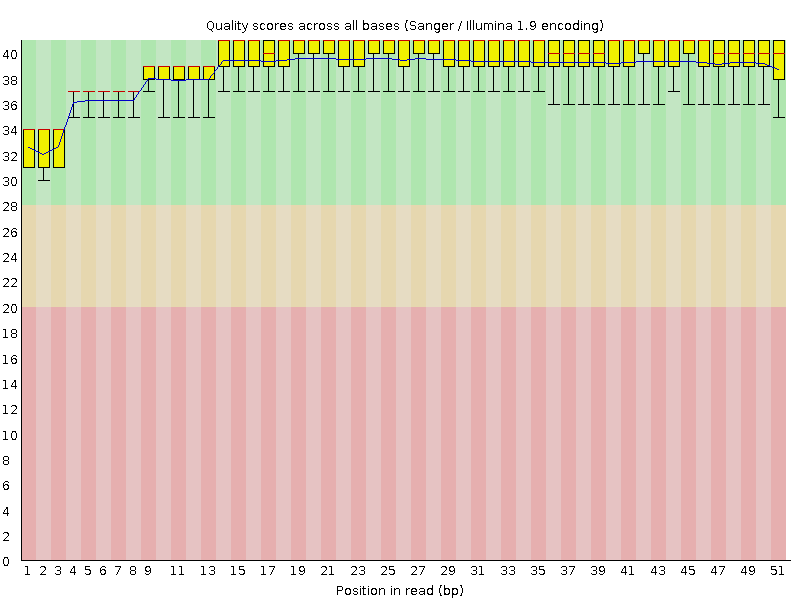
Trimming with TrimmomaticTrimmomatic with TRAILING:20 and MINLEN:50 didn't change the per base quality to any significant extent. What it did was to covert a check in the "Per tile quality" section into a warning:Before trimmming:  After trimmming:  Thus it was decided to work with the original set of reads instead of the trimmed ones. FastQC report after trimming There were 24156 reads in the original file and 24051 reads in the resulting file. Mapping the readsThe hisat2 program was launched without the --no-spliced-alignment parameter since we are dealing with transcriptome this time, where the reads can contain exon junctions.This was the program's output:
24156 reads; of these:
24156 (100.00%) were unpaired; of these:
502 (2.08%) aligned 0 times
23643 (97.88%) aligned exactly 1 time
11 (0.05%) aligned >1 times
97.92% overall alignment rate
Below there is a .sam file containing those 23654 reads mapped to the reference sequence (chr5.fasta):Alignment Alignment analysisThe binary file returned by samtools view: mapped_transcriptome.bamThe binary file returned by samtools sort: mapped_transcriptome_sorted.bam Indexed file produced by samtools index: mapped_transcriptome_sorted.bam.bai 23654 reads were mapped to the chromosome sequence, 502 were not. Additional information from hisat2 can be found above or in the .sam file produced by it (link in the previous section). Counting the reads with htseq-countThe htseq-count program counts how many reads map to each feature from the feature file (in this case features are genes). Some of its options are -f, -s, -i, -m. -f specifies the format of the input file (sam/bam, default is sam); -s {yes, no, reverse} establishes whether the data is from a strand-specific assay, 'reverse' means 'yes' with reverse strand interepretation; -i sets the feature ID for a GFF feature file (default is gene_id suitable for Ensembl GTF files); -m {union, intersection-strict, intersection-nonempty} specifies the protocol to follow in ambiguous mapping cases: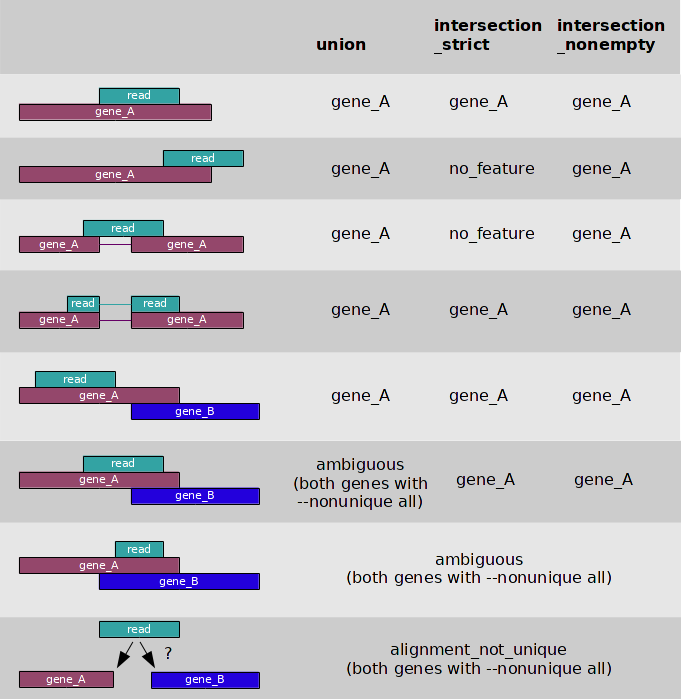 The resulting file contained a host of genes with zero reads mapped to each of them, so regular expressions were used to extract the genes with nonzero number of pertaining reads: ENSG00000181163.9 22529 ENSG00000249353.2 316 __no_feature 798 __not_aligned 502 __alignment_not_unique 22Here is the same data in a file. To sum up, out of 24156 reads 502 were no aligned, 798 were aligned but didn't match any genes, 22 were aligned ambiguously and were not resolved by the union mode, 22529 reads were aligned with the ENSG00000181163.9 gene, and 316 reads were aligned with the ENSG00000249353.2 gene. The ENSG00000181163.9 gene codes for nucleophosmin 1 (NPM1), which is a protein associated with nucleolar ribonucleoprotein structures and involved in the biogenesis of ribosomes. It binds single-stranded and double-stranded nucleic acids, preferentially G-quadruplex forming ones. ENSG00000249353.2 is nucleophosmin 1 pseudogene 27 Table with all the commands used
|
© Stanislav Tikhonov, 2018