1. Определение функции и таксономии нуклеотидной последовательности
Работа велась с полученнной в 6 практикуме консенсусной последовательностью. Так как мы ничего не знаем о последовательности и ищем ближайшие гомологи, имеет смысл использовать алгоритм blastn. Параметры поиска приведены в Таблице 1. Выдача сохранена в файле pr8.txt.
Судя по выдаче, наша последовательность является частичной кодирующей для гистона H3. Последовательность очень консервативная, поэтому, если она взята из организма определённого вида, у выравнивания должен быть стремящийся к ста процент индентичности и малое значение E-value. Поиск таких выравниваний немного осложняется тем, что в нашей последовательности много полиморфизмов, то есть процент идентичности будет ниже истинного.
Самые низкие значения E-value с самым большим процентом идентичности у Psolus phantapus Dendrochirotida sp.. Посмотрим раздел Distance tree of results. Поставим максимальную долю различий (Max Seq Difference) 0.1. Результат представлен на Изображении 1.
Судя по дереву, последовательность была взята как минимум у представителя таксона Psolidae, вероятнее всего рода Psolus. Выравниванием последовательностей рода Psolus между собой показало, что для представителей одного вида этого рода характерно различие в 4 нуклеотида. Похожее имеем в нашей последовательности. Следовательно, она скорее всего взята из представителя рода Psolus, предположительно Psolus phantapus.

2. Поиск генов белков в неаннотированной нуклеотидной последовательности
Внимание! Сейчас вы увидите, как я перебирал 5 организмов, примерно по 3 генома для каждого, по 3-5 контигов в каждом геноме. Чтобы нигде не оказалось никаких находок.

Теперь к делу. Кажется, находки выпадают только если брать, не что-то близкородственное к хорошо аннотированным геномам, но если брать какую-то далёкую непонятную хтонь [простите за стиль, я просто слишком устал]. Этот вывод я сделал после просмотра практикумов однокурсников. Наконец я нашёл сборку генома дрожжей (важно, что они мало изученные) Yarrowia divulgata [параллельно делал с Dipodascus geniculatus, получилось, но взял только одно]. Из неё взял 10 контиг длиной 98608 нуклеотидов, AC ULGQ01000010.1.
Для поиска генов сравним контиг с аннотированными последовательностями. Нам нужны совпадения не в нуклеотидах, а в аминокислотных остатках в последовательности белка, поэтому используем алгоритм blastx, транслирующий нуклеотидную последовательность контига и ищущий соответствия в белковой базе данных, в нашем случае в RefSeq Select proteins. Выдача BLAST сохранена в файле pr8_1.txt, параметры запуска BLAST указаны в Таблице 2.
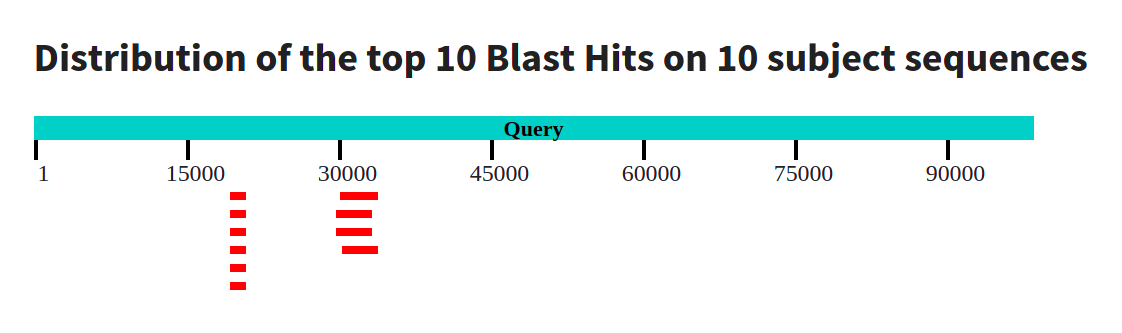
В окне Graphic Summary (Изображение 2) увидим положение находок относительно контига. Я решил выбрать для рассмотрения самую длинную последовательность, длины выравниваний от 1710 до 1828 позиций. Все находки с E-value 0.0, идентичность около 35%, позитивных позиций около 55%. Соответствующие нуклеотидные последовательности ложатся примерно на один и тот же участок и кодируют белки из семейства хромодомен-хеликаз-ДНК-связывающих белков мыши и человека. Из этого можно сделать вывод, что с большой долей вероятности мы нашли ген, кодирующий какой-то из хромодомен-хеликаз-ДНК-связывающих белков.
3. Интепретация карты локального сходства гомологичных хромосом двух бактерий
Для построения карты локального сходства я выбрал геномы Neisseria meningitidis и Neisseria lactamica. Далее использовали алгоритм megablast с длиной слова 64 и E-value 5e-101 (увеличивал, пока карта не очистилась от шума).
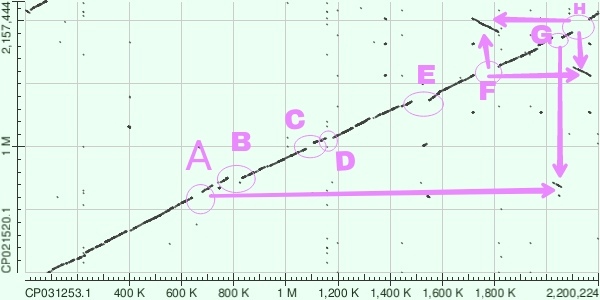
На Изображении 3 показана карта локального сходства. Описание первого участка буду производить со ссылками на горизонтальные координаты.
На участке 0-100 видим фрагмент с иллюзией транслокации. На самом деле это результат того, что мы рассматриваем кольцевой геном. Координаты просто сместились из-за того, что у двух последовательностей начало формально отмечено в разных точках. Далее рассмотрим A и G. Здесь произошла транслокация с инверсией. Для горизонтальной оси фрагмент переместился из A в G, в вертикальной он остался в А. Похожая ситуация в F и G. Оба фрагмента инвертировались и поменялись друг с другом местами. В участках B, C, D, E видим индели с дополнительными мелкими изменениями, которые по карте восстановить слишком сложно.