Выравнивание последовательностей
Сначала я скопировала последовательности для выравнивания в файл shortseqs.fasta, чтобы затем импортировать в GeneDoc и, соответсвенно, выравнять. Результат выравнивания можно увидеть в документе alignment1.msf, либо на этом изображении:
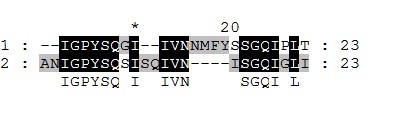
Как видно, часть аминокислотных остатков совпадают. Есть несколько способов более или менее хорошо выровнять эти последовательности (например, можно заметит схожие остатки M и I), то в таких случаях количество и расположение гэпов сводит на нет все положительные изменения и самым адекватным выравниванием является такое (по матрице BLOSUM62).
Теперь построим карту локального сходства этих последовательностей. Результаты задания находятся в файле map.xlsx на первом листе. Единицей отмечены полностью совпадающие остатки, цифрой в скобочках - сходство остатков в матрице BLOSUM62. Цветом выделены ячейки, обозначающие путь выравнивания. Там где они идут по диагонали ровно вниз вправо, выравнивание проходит без перерывов и подряд; там, где есть разрывы в этой диагонали, соответсвенно, вставлены гэпы. Соответствие остатка гэпу показано в карте розовым цветом.

Теперь, пользуясь программой bl2seq, выровняем первый фрагмент из предыдущих заданий с последовательностью моего белка YABJ_BACSU (1QD9). Если выравнивать последовательности при параметрах по умолчанию, мы видим следующий результат:
>lcl|37901 unnamed protein product
Length=23
Score = 52.0 bits (123), Expect = 1e-15, Method: Composition-based stats.
Identities = 23/23 (100%), Positives = 23/23 (100%), Gaps = 0/23 (0%)
Query 14 IGPYSQGIIVNNMFYSSGQIPLT 36
IGPYSQGIIVNNMFYSSGQIPLT
Sbjct 1 IGPYSQGIIVNNMFYSSGQIPLT 23
Мы видим, что первая последовательность представляет собой часть второй (с 14го остатка по 36й).
Теперь выровняем последовательность моего белка с последовательностью гомологичного ему.
Первый белок - YABJ_BACSU из Bacillus subtilis;
Второй белок - MUG71_SCHPO из Schizosaccharomyces pombe.
Результаты выравнивания при параметрах по умолчанию здесь.
Как мы видим, есть два участка сходства.
Первый - 49-105 (326-383 во втором белке), процент идентичности - 41%, сходства - 59%, один гэп (2%), который представляет собой, очевидно, один разрыв.
Второй - 10-38 (417-445 во втором белке), процент идентичности - 62%, сходства - 69%, гэпов нет совсем, разрывов, соответственно, тоже.
Карта: 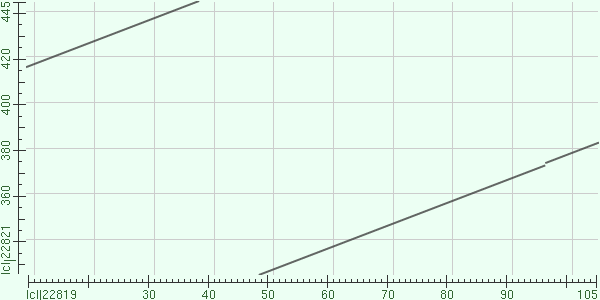
Дополнительное задание:
Попробуем выравнять те же последовательности при других параметрах. В первый раз мы изменим матрицу сходства на PAM70 (что-то, что менее всего похоже на исходную матрицу)), во второй - установим "ценность" гэпа на "Existence: 8 Extension:2". Целиком результаты выравниваний (первого и второго) здесь приводить не буду, их можно скачать по указанным ссылкам. Сравним области сходства всех трех выраниваний, включая самое первое при исходных настройках (назовем его нулевым).
Первый участок:
0: + +NL +L G S + V TV ++ M +FAE N VY +YFD T+ P+RSCV 1: +NL L G S V TV ++ M FAE N VY YFD T+ P+RSCV 2: + +NL +L G S + V TV ++ M +FAE N VY +YFD T+ P+RSCV
Процент идентичности 0 с 1 - 88,23%, 0 с 2 - 100%.
Второй участок:
0: APA IGPYSQ I N + + SGQI L PS 1: ++ APA IGPYSQ I N + SGQI L PS 2: APA IGPYSQ I N + + SGQI L PS ++ + ++ ++ L+ A A
Процент идентичности 0 с 1 - 86,36%, 0 с 2 - 64,51%.
Как мы видим, изменение параметра учета гэпов несильно повлияло на результаты, так как самих гэпов было достаточно мало.
Дополнительное задание:
Результаты этого задания находятся в том же файле map.xlsx на втором и третьем листе. Для заполнения матрицы использовались формулы, которые можно там же и посмотреть.