Анализ результатов поиска по профилю
Разделение выравнивания представителей домена на две группы
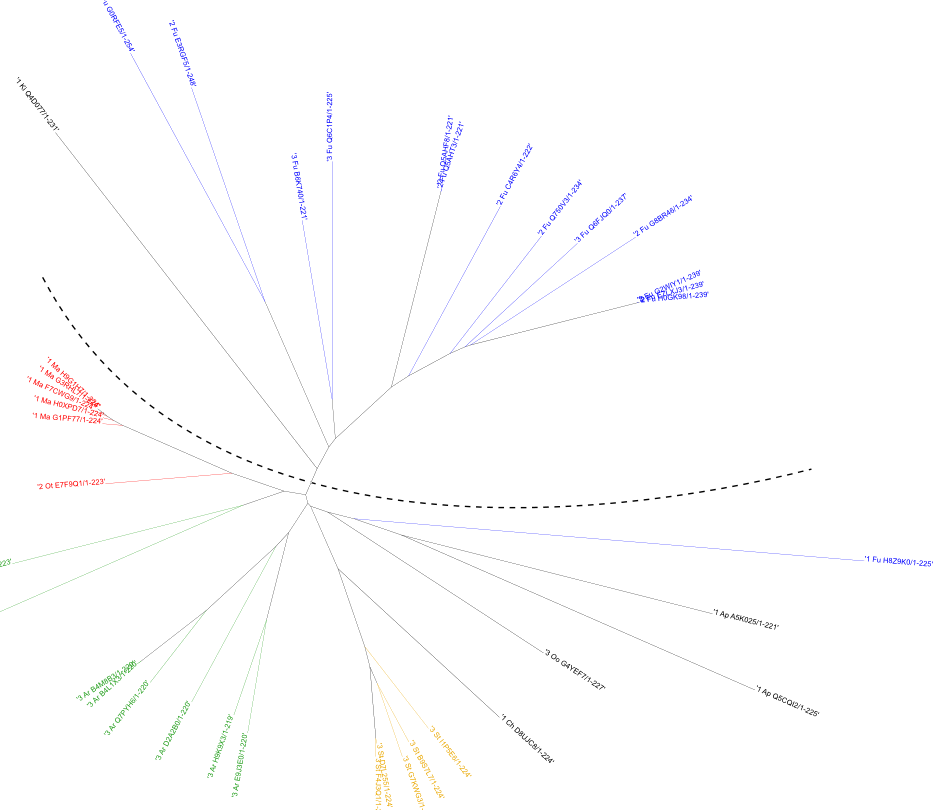
Напомню, что изначально было выбрано три архитектуры для анализа: ATP_bind 4, ATP_bind 4 + Ribonuc_L-PSP и ATP_bind 4 + Ribonuc_L-PSPx2. Выравнивания домена в группах каждой из трех архитектур довольно похожи (с некоторыми различиями, конечно), поэтому я сомневалась, что профиль, построенный на основе выбора одной архитектуры, будет обладать хорошими специфичностью и чувствительностью. Поэтому я решила разделить последовательности на две группы, опираясь на построенное филогенетическое дерево. Разделение показано на рисунке ниже (как видно, оно не совсем соответствует разделению по таксономии):
Построение профиля, отличающего одну группу от другой
Последовательности были заново распределены по группам, согласно новому выбору (jar-файл).
Профили были построены по обеим группам, чтобы выбрать лучший. Первый, второй. Этап нормализации был опущен.
По score-кривым и даже просто по соответствиям находок и групп (vlookup-колонка) видно, что первый профиль выглядит лучше второго. У него можно определить порог, который достаточно хорошо (только одна ошибка) разделяет последовательности из двух групп - около 57.7.
Была построена ROC-кривая для первого профиля. По ней видно, что порог действительно выбран правильно. Таким образом, удалось создать профиль, позволяющий достаточно эффективно отличить заданные группы последовательностей.