Сборка генома de novo
С помощью кода доступа SRR4240379 я скачала fastq-файл проекта по секвенированию бактерии Buchnera aphidicola.
1. Подготовка чтений программой trimmomatic
Прежде всего был скачан архив с прочтениями, затем распакован командой gunzip в папку /nfs/srv/databases/ngs/ulyana/pr15.
Все адаптеры для Illumina из файлов в директории /P/y15/term3/block4/adapters были объединены в один файл adapters.fasta командой
cat /P/y15/term3/block4/adapters/*fa >> /nfs/srv/databases/ngs/ulyana/pr15/adapters.fasta
.
Далее командой
java -jar /usr/share/java/trimmomatic.jar SE -phred33 SRR4240379.fastq srr_out.fastq ILLUMINACLIP:adapters.fasta:2:7:7
были удалены возможные остатки адаптеров.
С концов чтений были удалены плохие буквы, оставлены только чтения не менее 30. Программа
java -jar /usr/share/java/trimmomatic.jar SE -phred33 srr_out.fastq trimm.out TRAILING:3 MINLEN:30
.
принимает на вход файл srr_out.fastq, полученный на предыдущем этапе,
удаляет прочтения короче 30 (MINLEN:30) и удаляет нуклеотиды ниже качества "3" с конца прочтений (TRAILING:3).
В результате получен файл trimm.out.
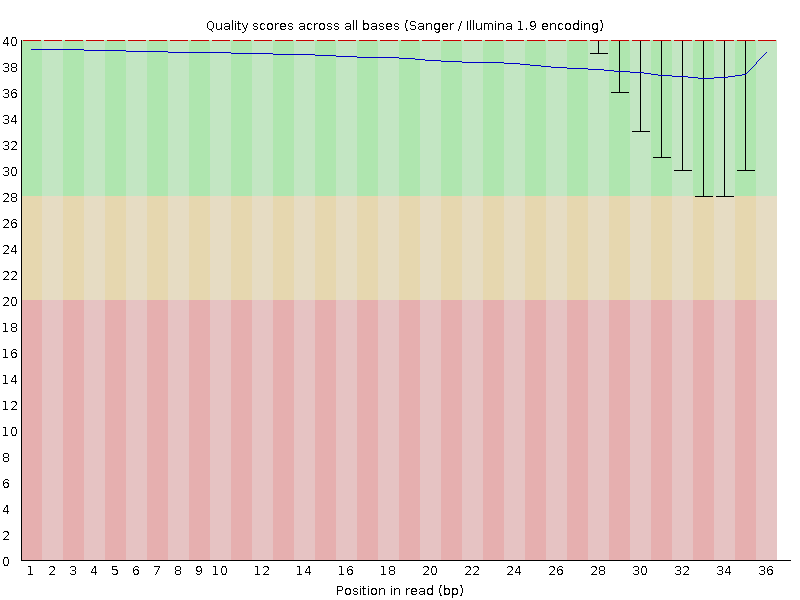
Чтобы сравнить качество прочтений до и после очистки, я использовала команду fastqc, в результате работы которой были получены 2 html страницы:
SRR4240379_fastqc.html и trimm.out_fastqc.html.
После очистки осталось 6993284 чтний из 7400155 изначальных. Результаты представлены на рис. 1 и 2. Видно, что качество чтений улучшилось.
|
|
|
Рис. 1 Per base sequence quality до очистки |

|
|
|
Рис. 2 Per base sequence quality после очистки |
2. Работа с программой velveth
Для подготовки k-меров длиной 29 была использована команда
velveth k_mer 29 -fastq -short trimm.out,
где k_mer - название папки для записи выходных файлов. Длина k-меров равно 29, -short задает, что чтения короткие и непарные, -fastq, что входные файлы задаются в соответствующем формате.
Входной файл trimm.out содержит очищенные чтения.
В результате, в директории k_mer оказались результаты работы программы.
3. Сборка на основе k-меров с помощью программы velvetg
Данная программа строит и анализирует граф де-Брёйна (как и предыдущая - velveth), где k-меры играют роль вершин.
Контиги на основе 29-меров были собраны командой velvetg k_mer.
Получено 2 файла: contigs.fa с последовательностями контигов и stats.txt, со статистикой.
Всего было найдено 974 контигов. N50 составляет 31053 (прочтением такой длины можно покрыть больше половины генома). Информация о 3-х самых длинных контигах представлена с таблице №1.
Если считать типичным покрытие равное 9 (медиана), то аномально больших покрытий довольно много. В таблице № 2 приведены несколько таких примеров.
Также есть несколько контигов со значением покрытия inf, что, вероятно, является результатом ошибки работы программы.
Аномально малых покрытий не было обнаружено.
| Таблица №1 Информация о самых длинных контигах |
||
| ID | Длина | Покрытие |
| 5 | 82103 | 47,938394 |
| 2 | 70497 | 49,611544 |
| 10 | 43532 | 50,862607 |
| Таблица №2 Информация о контигах с аномально большим покрытием |
||
| ID | Длина | Покрытие |
| 890 | 1 | 643980 |
| 55 | 278 | 220,661871 |
4. Анализ
Для трех самых длинных контигов был запущен megablast с хромосомой бактерии Buchnera aphidicola (GenBank/EMBL AC — CP009253). Выравниваяния имею хорошие показатели (таблица №3).
| Таблица №3 Характеристика выравниваний некоторых контигов с заданной хромосомой |
||||||
| ID контига | Контиг | Расположение на хромосоме | Query cover | Identity | E value | Выравнивание |
| 5 | con5.fa | 467412-474667 500370-508806 510438-516539 523105-528679 462496-467421 481997-488106 474844-480660 517766-521500 496111-500325 451729-454069 |
69% | 77% | 0.0 | al_con5.txt |
| 2 | con2.fa | 528977-550219 550361-555887 573092-582686 563837-567489 571558-57307 584329-587055 561741-563423 593743-594099 |
65% | 81% | 0.0 | al_con2.txt |
| 10 | con10.fa | 11103-2004 620926-613658 604795-599832 627104-621055 613671-611633 14229-13994 611524-611229 |
68% | 78% | 0.0 | al_con10.txt |
| 55 | con55.fa | - | 64% | 83% | 0.012 | al_con55.txt |
Что касается аномально длинных контигов, то я сделала выравнивание только одного из них, 55-го, поскольку для другого это бессмысленно, учитывая его длину. Однако, алгоритмы megablast и даже discontiguous megablast не нашли достаточно сходных последовательностей во всем геноме, чтобы построить выравнивание. Поэтому я использовала алгоритм blastn. Характеристики полученного выравнивания также представлены в таблице №3. Как видно, значение e value слишком высокое, чтобы считать выравнивание достоверным. Вероятно, покрытие такое большое из-за малой длины контига: высока вероятность случайного соответствия. По этой же причине я не привожу координаты данного контига на хромосоме.