Ресеквенирование.Поиск полиморфизмов у человека.
Картирование чтений на референсный геном.
Для картирования чтения была использована следующая команда:
bwa mem -t 10 ./bwa/chr3.fna ./paired_1.fastq.gz ./paired_2.fastq.gz > reads.sam 2> mem_log.txt
Параметры, использующиеся в этой команде:
Анализ sam-файла с выравниванием.
В sam-файле с выравниванием можно выделить две "части" - заголовки с тэгами и саму таблицу. Строки заголовков отличаются от строк таблицы наличием символа "@" в начале строки. В
заголовках есть краткая информация о программе, с помощью которой производилось картирование, чтениях и референсном файле. В теле файла содержится информация о качестве секвенирования и
картирования для каждого рида.
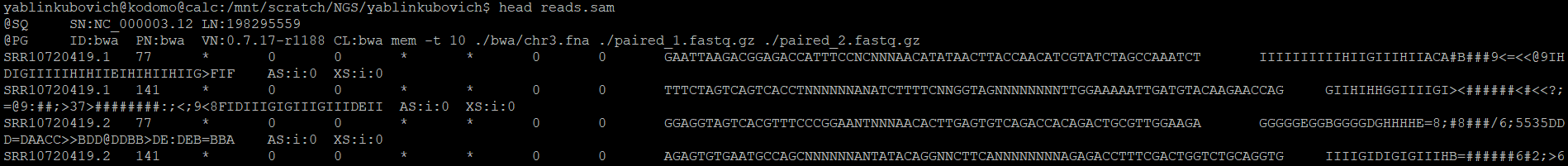
Информацию о содержимом первых 11 столбцов таблицы файла reads.sam можно посмотреть в таблице 1.
| Столбец | Описание |
| QNAME | ID рида |
| FLAG | Характеристика выравнивания в виде суммы нескольких других характеристик (не очень понял, в википедии мало информации, а ссылка с википедии ведёт на сайт Decoding SAM flags) |
| RNAME | Название референсной последовательности, с которой выровнялся рид |
| POS | Вычисляется по формуле (1-(самая левая позиция выравнивания)) |
| MAPQ | Качество картирования |
| CIGAR | Строка с информацией о выравнивании чтения с референсом |
| RNEXT | Референсное название места картирования следующего рида в образце |
| PNEXT | Позиция, на которую картировался следующий рид |
| TLEN | Расстояние между крайними точками выравнивания |
| SEQ | Последовательность рида |
| QUAL | Phred-score каждого нуклеотида в риде |
Также в таблице есть ещё опциональные столбцы:
и так далее.
Вес файла reads.sam приблизительно равен 16 Гб.
Получение и индексирование bam файла.
Полученный sam-файл был отсортирован в bam файл командой:
samtools sort -@ 10 -o read_mapped.bam reads.sam 2> bam_log.txt
Здесь -@ 10 - опция, задействующая 10 ядер для ускорения процесса. Опция -о определяет название файла на выходе, а ошибки перенаправляются в файл
bam_log.txt
Полученный файл весит примерно 5 Гб - почти в три раза меньше, чем несортированный.
Далее полученный файл был проиндексирован с помощью samtools index:
samtools index -@ 10 read_mapped.bam
Анализ bam файла
Для того, чтобы можно было просмотреть статистику по файлу bam, была выполненая следующая команда:
samtools flagstat -@ 10 read_mapped.bam > flagstat.txt
Полученный файл можно посмотреть здесь.
Из него можно получить следующую информацию:
Картировано всего 13,78% чтений (информация в строке mapped), картированы в корректных парах чтений 10.84% чтений (в строке paired properly). Проценты отличаются, потому что риды могут
картироваться на геном несколько раз, и тогда чтения одной пары могут быть далеко друг от друга.Малый процент картированных чтений можно объяснить тем, что мы картируем чтения
всего экзома только на одну хромосому.
Получение картированных чтений.
Сначала, для того, чтобы узнать название хромосомы, выполним команду:
samtools faidx chr3.fna -o chr3.fai
Для получения из общего файла чтений, картированных только на 3-ю хромосому, и дальнейшего конвертирования в bam-файл были выполнены следующие команды:
samtools view -h read_mapped.bam NC_000003.12 > read_chr3.sam
samtools view -b read_chr3.sam > read_chr3.bam
После этого для просмотра статистики была использована команда:
samtools flagstat -@ 10 read_chr3.bam > flagstat_chr3.txt
Файл можно посмотреть здесь.
Картировано уже 88.86 процентов чтений
(10961608), 8617706 чтений (70.14%) картированы в корректных парах. Можно видеть, что численное количество чтений не изменилось, однако заметно выросло процентное соотношение из-за того,
что мы теперь картируем чтения не всего экзома, а только третьей хромосомы.
Для получения только правильно картированных пар чтений мы воспользовались командой:
samtools view -@ 10 -f 0x2 -bS read_chr3.bam > read_chr3_cor.bam
Затем для получения статистики:
samtools flagstat -@ 10 read_chr3_cor.bam > flagstat_chr3_cor.txt
Файл можно посмотреть здесь.
В нем содержится следующая информация:
Картированных чтений - 8619870 (100%)
Правильно картированных пар чтений - 8617706 (100%)
Как видно из результатов, теперь все чтения картированы. Это очень хорошо
Далее файл был проиндексирован с помощью команды:
samtools index -@ 10 read_chr3_cor.bam
Подготовка выравнивания к поиску вариантов.
Дублированные чтения были маркированы с помощью следующей команды:
picard MarkDuplicates -M metrix.file.txt -I read_chr3_cor.bam -O marked_chr3.bam
К полученному файлу применили команду flagstat:
samtools flagstat -@ 10 marked_chr3.bam > marked_flagstat.txt
Файл можно посмотреть здесь. Из полученного файла видно, что нашлось 646626 дублированных
чтений.