Филогенетические деревья. Занятие 4.
1. Построение дерева по нуклеотидным последовательностям
Данный блок содержит описание хода работы, для просмотра результатов
перейти сюда
В предыдущих заданиях (ссылки: занятие 2, занятие 3)
использовалось простроение деревьев по аминокислотным последовательностям
белков. Но часто для исследования филогении применяется построение
по нуклеотидным последовательностям, особенно - по последовательностям
рибосомальных РНК (медленно эволюционируют, имеют гомологи во всех организмах, соблюдаются (?) молекулярные часы).
В частности, в текущей работы выбраны последовательности 16S рРНК фирмикут.
Этапы работы.
1. Скриптом shell с запросами к entret получила записи о белках
HSLO (не принципиально, какой белок брать на этом этапе) выбранных
бактерий из предыдущих занятий.
Напоминание как запускать скрипты в shell:
chmod +x script.sh
./script.sh
Напоминание как формулировать запрос к entret банку Swiss-Prot:
entret sw:hslo_bacan -out hslo_bacan.entret
2. В файлах вручную нашла ссылки на записи EMBL:
BACAN AE016879
BACSU D26185
LISMO AL591974
GEOKA BA000043
STRP1 AE004092
LACAC CP000033
STAA1 AP009324
STRPN AE005672
3. Скриптом shell с запросами к entret получила данные записи.
Напоминание как формулировать запрос к entret банку EMBL:
entret embl:AE016879 -out AE016879.entret
4. Вручную произвела поиск проаннотированных 16S rRNA (поле FT - "rrna"). Получила список:
BACAN AE016879 9335 10841
BACSU D26185 73411 74963
LISMO AL591974 37466 39020
GEOKA BA000043 10421 11913
STRP1 AE004092 17170 18504
LACAC CP000033 59255 60826
STAA1 AP009324 531922 533476
Обратить внимание: все последовательности расположен на прямой цепи ДНК.
В файле, cоотвтствующем записи о STRPN (Streptococcus pneumoniae) не оказалось аннотации рРНК.
5. Скриптом python с запросами к seqret (опция -sask) получила fasta-файлы с последовательностями 16S rRNA.
6. С помощью blastn нашла последовательность 16S rRNA в геноме STRPN (Streptococcus pneumoniae).
Как это сделать: получить последовательность генома STRPN в fasta-формате,
сформировать базу данных для blast-a,
запустить поиск по ней с запросом последовательностью 16S rRNA близкородственной бактерии
(STRP1 в данном случае, смотри дерево отобранных бактерий).
Получение генома STRPN:
seqret embl:AE005672 -out STRPN.fasta
Формирование базы для blast:
makeblastdb -in STRPN.fasta -out STRPN -dbtype nucl
Запрос к blastn:
blastn -db STRPN -task blastn -out STRPN_result.txt -evalue 0.001
-outfmt 7 -query 16S_STRP1.fasta
Содержание выходного файла с номерами совпадений (т.е. 16S rRNA STRPN):
# BLASTN 2.2.26+
# Query: AE004092 AE004092.1 Streptococcus pyogenes M1 GAS, complete genome.
# Database: STRPN
# Fields: query id, subject id, % identity, alignment length, mismatches, gap opens, q. start, q. end, s. start, s. end, evalue, bit score
# 4 hits found
AE004092 AE005672 94.76 1336 68 2 1 1335 15450 16784 0.0 2087
AE004092 AE005672 94.76 1336 68 2 1 1335 1814956 1813622 0.0 2087
AE004092 AE005672 94.76 1336 68 2 1 1335 1913613 1912279 0.0 2087
AE004092 AE005672 94.76 1336 68 2 1 1335 1975629 1974295 0.0 2087
# BLAST processed 1 queries
Обратить внимание: последовательность расположена на прямой цепи ДНК.
7. Получила последовательность fasta 16S_STRPN, скриптом python сформировала файл .fasta со всеми полученными 16S rRNA с сокращенной мнемоникой.
Ссылка на этот файл: genomes.fasta
8. Запустила на kodomo muscle, получила выравнивание. (Проще, правда, сразу выполнить это в MEGA... см. следующий пункт.)
Cсылка на него: genomes.afa.
Напоминание как это сделать:
muscle -in genomes.fasta -out genomes.afa
9. Выравнивание открыла с помощью MEGA, указала Analyse, построила дерево в меню Phylogeny, метод ME (Minimum Evolution).
Можно было открыть в MEGA сам .fasta и выровнять с помощью muscle здесь.
Внимание! Промежуточные результаты всех этапов работы, а также использованные скрипты можно найти в рабочей директории
"agalicina/Term4/Block1/Practice4/1"
Результаты работы.
Таблица AC и координат 16S рРНК в них:
| Мнемоника | AC | Начальная координата | Конечная координата | Цепь ДНК |
| BACAN | AE016879 | 9335 | 10841 | прямая |
|
LISMO | AL591974 | 37466 | 39020 | прямая |
|
GEOKA | BA000043 | 10421 | 11913 | прямая |
|
STRP1 | AE004092 | 17170 | 18504 | прямая |
|
LACAC | CP000033 | 59255 | 60826 | прямая |
|
STAA1 | AP009324 | 531922 | 533476 | прямая |
|
BACSU | D26185 | 73411 | 74963 | прямая |
|
STRPN | AE005672 | 15450 | 16784 | прямая |
Выравнивание получено с помощью muscle:
genomes.afa.
Метод реконструкции: Minimum Evolution, встроенный в MEGA.
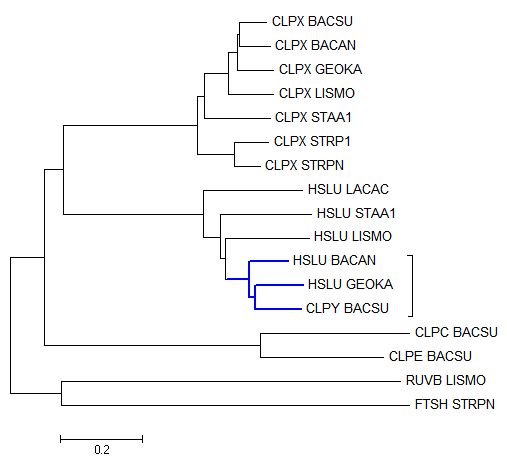
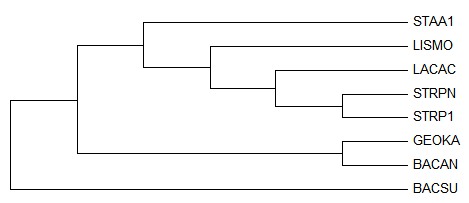
Полученное дерево:
Короткий анализ: по сравнению с исходным деревом неправильно прикреплен лист GEOKA, неверна ветвь
{BACAN,BACSU,GEOKA,STAA1}vs{LISMO,LACAC,STRP1,STRPN}.
Ошибки с теми же листьями были характерны и для построений по белку HSLO в предыдущих заданиях. Для
более точного ответа хорошо бы восстановить дерево по 16S рРНК разными методами.
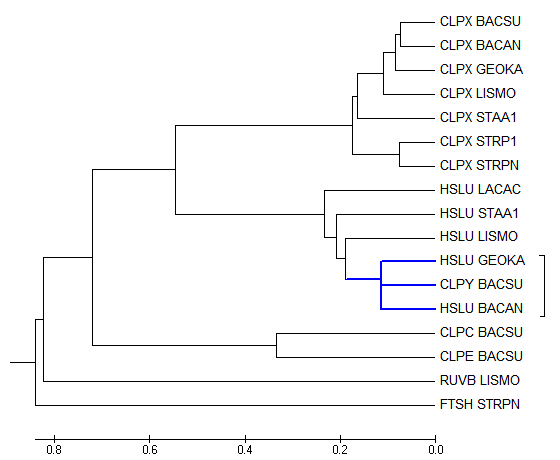
Результат NJ (такой же):
Результат MP:
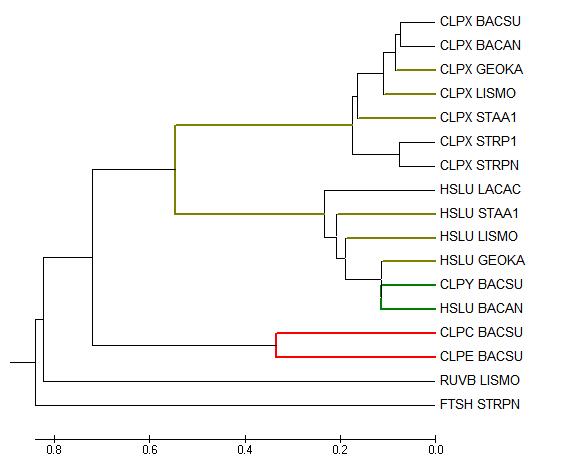
Результат UPGMA c бутстрепом:
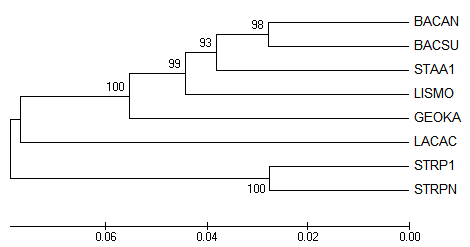
Ошибки в прикреплении всё тех же листьев GEOKA и STAA1/LISMO, UPGMA вообще прикрепляет GEOKA выше всех Bacillales, отдельно от других Bacillaceae.
Можно сделать вывод, что для достоверной реконструкции выборка слишком мала, потому что даже восстановление по 16S рРНК дает значительные ошибки реконструкции.
2. Построение и анализ дерева, содержащего паралоги
Данный блок содержит описание хода работы, для просмотра результатов
перейти сюда
Среди гомологов белков выделяют ортологи и паралоги. Ортологи - гомологичные белки,
произошедшие в результате видообразования. Паралоги - произошедшие в результате дупликации генов.
Последнее означает, что в результате дупликации по какой-то
причине в геноме в разных местах появились две копии одного
гена и с этого момента эволюционируют
отдельно, находясь при этом в одном организме и кодируя схожие белки.
В исследуемой группе организмов можно найти все гомологи данного белка, затем построить дерево, в котором будут отмечены названия белков и виды, из которых они произошли.
Проанализировав последнее, выясняют историю эволюции белков: дивергенцию и дупликации.
В текущей работе предлагалось найти в
заданном файле proteo.fasta все достоверные гомологи белка CLPX_BACSU, затем выровнять их последовательности и построить дерево гомологов. Приняв реконструкцию верной, предлагалось проанализировать и выяснить, сколько найдено пар ортологов, пар гомологов.
Этапы работы.
1. В предложенном файле proteo.fasta, взятом с диска P kodomo, находятся записи
UNIPROT, относящиеся к белкам из предложенной ещё на
первом
занятии выборки фирмикут.
По нему создаю базу данных для blastp:
makeblastdb -in proteo.fasta -out proteo -dbtype prot
2. С помощью blastp провожу поиск гомологов CLPX_BACSU (заранее получен .fasta с помощью seqret):
blastp -query clpx_bacsu.fasta -db proteo -out blastp.txt -evalue 0.001 -outfmt 7
3. Из этого файла
blastp.txt
получаю белки только
выбранных
раньше бактерий.
Ссылка на полученный список, он иногда содержит несколько хитов с одним белком.
4. С помощью запросов к seqret скриптом shell получаю .fasta всех
найденных белков. С помощью скрипта python переписываю эти
последовательности, но с укороченной мнемоникой вида "белок_организм", в один файл.
Результат:
proteins.fasta
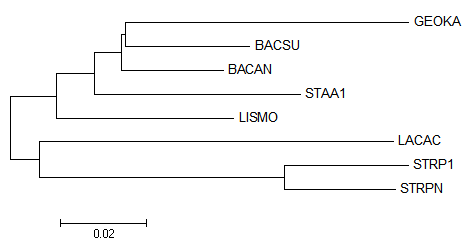
5. Далее выравниваю с помощью muscle, например, сразу в MEGA.
Сторою дерево, например, NJ и UPGMA.
Внимание! Промежуточные результаты всех этапов работы, а также использованные скрипты можно найти в рабочей директории
"agalicina/Term4/Block1/Practice4/2"
Главная страница
Страница семестра
© Галицына Александра, 2012