EMBOSS
Упражнения
- Команда:
seqretsplit
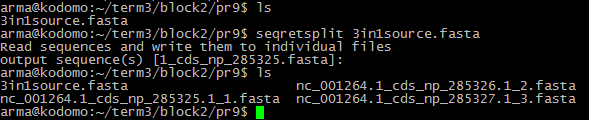
Файл с результатом:
nc_001264.1_cds_np_285325.1_1.fasta
nc_001264.1_cds_np_285325.1_1.fasta
nc_001264.1_cds_np_285325.1_1.fasta
- Команда:
seqret
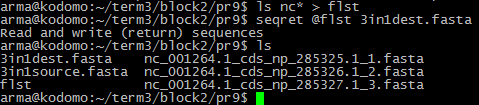
Файл с результатом:
3in1dest.fasta
- Команда:
transeq (трансляция по указанной таблице)
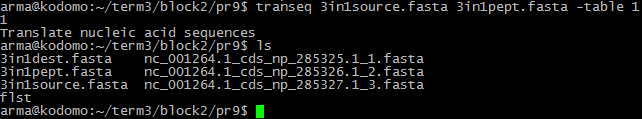
Файл с результатом:
3in1pept.fasta
- Команда:
transeq(трансляция всех возможных ORF)
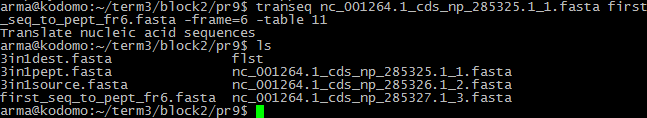
Файл с результатом:
first_seq_to_pept_fr6.fasta
- Команда:
cusp
Файл с результатом:
first_seq_codons_freqs
Трансляции открытых рамок посредством EMBOSS
Организм - Deinococcus radiodurans, часть генома - вторая хромосома (NC_001264.fasta)длиной 412,348 Кб и содержащая по аннотации 368 генов белков и 1 ген тРНК.
Рамки получены командой getorf NC_001264.fasta -minsize 180 -circular yes -table 11 и выведены в файл nc_001264.orf
Длины коордиаты и идентификаторы рамок получены командой infoseq nc_001264.orf -only -name -length -description -outfile nc_001264.info и сохранены в nc_001264.info
Для конвертации в xlsx с помощью инструмента "замена" (notepad++) убраны описания хромосомы и квадратные скобки с тире возле координат заменены на пробелы(nc_001264_info_spaceOnly).
После переноса столбцов в xlsx потребовались следующие манипуляции:
- Добавлен столбец ori, формируемый по условию: если from > to, ставим -1, иначе ставим 1. Данные вычислений скопированы в столбец, не зависящий от формулы.
- Сформированы два столбца, один из которых содержит минимальное между from и to значение, а второй - максимальное. Результаты вычислений этих столбцов также скопированы в независимые столбцы.
- Все зависимые столбцы (исходные from и to, формульные max, min и ori) удалены, на их место поставлены соответствующие данные независимых столбцов
- Данные упорядочены по возрастанию столбца from
- Добавлен столбец length (NC+stop), заполняемый по формуле (length*3+3), поскольку длина рамки выражена в АК-остатках, а для сравнения с аннотацией нам потребуется длина в нуклеотидах (*3) плюс неучтённый стоп-кодон (+3)
- Для уверенности в том, что после перестановок и прочих операций сохранилось соответствие между столбцами, добавлен "столбец контрольной суммы" test length: значения получаются по формуле (from - to) + 4 и должны совпадать со значениями length (NC+stop). "+4" - поскольку from считается не включительно (минусуется один нуклеотид) и не учтены стоп-кодоны (ещё минус 3 нуклеотида)
Результат: NC_001264.1_infoseq.xlsx.
Список аннотированных белков с NCBI
Доступен в виде XML-таблицы и FASTA-файла. Поскольку в оригинальной аннотации направление гена указывалось как plus и minus, а столбец length отсутствовал, пришлось:
- создать дополнительный столбец
- условием ЕСЛИ перевести значения plus/minus столбца origin в 1/-1 в новом столбце
- вставить независимый от формул столбец ori и скопировать туда вычисления
- выкинуть ненужный уже столбец origin и формулозависимый столбец
- заполнить столбец length вычитанием from из to.
Сравнение аннотированного и ORF
Гены, полученные с помощью поиска ORF, в среднем гораздо короче, чем аннотированные, и если построить график их следования друг за другом, они будут многократно накладываться (рыжая линия на рисунке). Аннотированные гены идут в основном не пересекаясь, даже с промежутками (синяя линия). Пояснения по графику: построен средствами Excel путём размещения значений from и to для одного гена на одной же высоте - 1 или 0 (или 1 или 2 для ORF)(значения from и to скомпонованы из двух столбцов в один друг за другом так, как они следуют в двух столбцах: A1,B1,A2,B2,A3...).
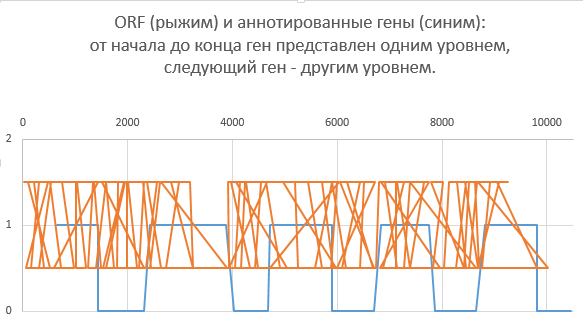
Они длиннее, чем ORF, и их гораздо меньше - соотношение примерно 1 к 10:
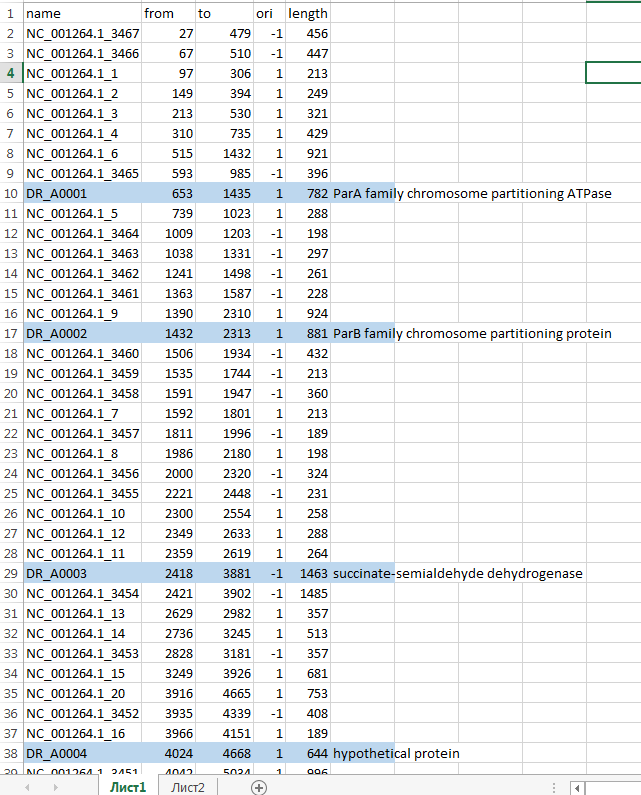
Сопоставление в Excel выполнено цветовым выделением ячеек с аннотированными генами, добавлением полученных ORF в ту же таблицу с отслеживанием соответствия столбцов и упорядочиванием по столбцу from.
Результаты сравнения в Excel
|