| Главная страница | Обучение | Обо мне | Ссылки |
Поиск сигналов | |||
|
Задание 1. Сравнение систем рестрикции-модификации, закодированных в двух штаммах одной бактерии Для сравнения систем рестрикции-модификации был взят геном бактерии Akkermansia muciniphila (Fasta-файл с последовательностью всего генома был взять с сайта NCBI: CP001071). Это грамм-отрицательная живущая в слизистой оболочке человеческого кишечника бактерия, котороая может использовать муцин в качестве источника углерода и азота. Недавние исследования показали, что она укрепляет барьер между собственно организмом и микроорганизмами кишечника, а также их метаболитами, которые при попадании в организм могут приводить к воспалительным процессам. В геноме Akkermansia muciniphila и в контигах, собранных для этой бактерии из метагенома человеческого кишечника, был проведен поиск известных сайтов рестрикции-модификации по методу Карлина (на специальном веб-сервисе). Полученные результаты приведены в excel-файле sites_RM.xlsx на листе whole genome. Среди возможных сайтов рестрикции избегаемыми оказались 12 (выделены голубым в таблице Excel), имеющие контраст меньше 0,78. Они вынесены на отдельный лист sites. После аналогичной процедуры поиска сайтов рестрикции в наборе контигов Akkermansia muciniphila (результаты приведены на листе contigs) были обнаружены 8 сайтов, имеющих контраст ниже 0,78, они выделены салатовым на листе sites. По найденным сайтам была сделана сводная таблица (лист sites). Анализируя полученную таблицу, можно сказать, что больше половины сайтов представлено в обоих штаммах. Среди сайтов, найденных в геноме, встретилось 4 таких, которые не нашлись в последовательностях контигов (а именно, AGATCT, CTCGAG, GATC, GTMKAC), и наоборот, 1 сайт нашелся в контигах и не нашелся в едином геноме другого штамма (GTATAC). Таким образом, всего было обнаружено 13 различных сайтов рестрикции-модификации. При этом в метагеноме их было обнаружено меньше, чем в полном геноме. Это может быть связано с тем, что в контиги просто не попали некоторые сайты (в пользу этого довода говорит и разница длин последовательностей: метагеном почти в 5 раз короче полного генома), либо действительно некоторые системы рестрикции-модификации были утеряны в ходе эволюции. Интересно, что в обоих штаммах есть сайт GCTAGC, имеющий контраст значительно выше 1 (конкретно - 1.636 в метагеноме и 1.239 в полном геноме). Это означает, что такое слово встречается в геномах чаще, чем ожидается. Уменьшенное количество систем рестрикции-модификации в штамме, обитающем в кишечнике, по сравнению с полным геномом другого штамма, можно объяснить тем, что, возможно, в кишечнике бактерия более защищена от воздействия бактериофагов и чужеродных плазмид, нежели в обычных условиях. Задание 2. Нахождение последовательности Шайна-Дальгарно Последовательность Шайна-Дальгарно (ШД)- это участок (последовательность нуклеотидов) мРНК у прокариот (бактерий и архей), расположенный перед инициаторным кодоном и необходимый для связывания мРНК с малой субъединицей рибосомы. Эта последовательность комплементарна 3'-концу 16S рРНК. Последовательность была впервые описана Дж. Шайном и и Л. Дальгарно в 1974 году[1]. Консенсус последовательности - AGGAGG. Со страницы моей археи Pyrobaculum oguniense TE7 была скачана полная последовательность хромосомы CP003316, а также таблица cо свойствами, которая была обработана в скрипте features2CDs.py для получения итоговой Excel-таблицы CDs.xlsx по всем кодирующим областям. Из таблицы были выбраны "хорошие" гены: длина не меньше 300 и не больше 1500 п.н. (здесь пригодился Обзор протеома археи Pyrobaculum oguniense TE7 из первого семестра, в котором есть гистограмма распределения длин белков, показавшая, что основная часть белков имеет длину 100-400 амк, т. е. около 300-1200 пн в CDs), были удалены гены, содержащие в описании продукта слова hypothetical, putative, uncharactirized и unknown. Всего генов было 2800, "хороших" осталось 971. Поскольку мотив ШД довольно короткий, необходимо ограничить области поиска в геноме последовательностей, действительно являющихся ШД, чтобы не спутать их с частью, например, гена. Для этого затем нужно было добавить координаты области поиска в таблицу описания генов. Это было сделано с помощью функции Excel ЕСЛИ с учетом направления гена. Были взяты области длиной 16 пн перед началом CDs. Итоговый файл с информацией об областях поиска ШД: в формате таблицы Excel и в файле с расширением txt (для скрипта). Для получения самих областей поиска был использован скрипт fragments2fasta.py (команда python fragments2fasta.py -f complete_genome.fasta). Выходной файл с короткими областями поиска: fragments.fast. Последовательности Шайна-Дальгарно были найдены с помощью сервера МЕМЕ: сначала на "хороших" генах (971 шт.), затем по всем генам. Параметры (Рис. 1): длина мотива от 4 до 10 нуклеотидов, поиск 3 различных мотивов.
Полученные 3 мотива последовательности Шайна-Дальгарно приведены на рис. 2.
Из полученных трех мотивов первый наиболее похож на консенсусную последовательность Шайна-Дальгарно, и он обладает наименьшим E-value - 1.2e-007. Кроме того, посмотрев окружение этого мотива (во вкладке More), можно обнаружить консенсусную последовательность ШД в чистом виде во многих областях поиска.
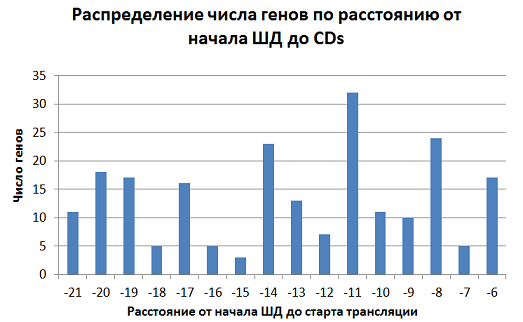
Далее первый мотив был отправлен в FIMO, чтобы найти его во всем геноме Pyrobaculum oguniense TE7. Предварительно upstream-область поиска была расширена до 21 нуклеотида. Файл с областями поиска по всему геному: fragments2.fasta. Порог по p-value: 0,001. Результаты получились не очень удовлетворительные: для 2800 генов всего в 217 случаях (т.е. для 7,75% генов) нашелся мотив, похожий на последовательность Шайна-Дальгарно. Ссылка на файл с выдачей FIMO: fimo.xlsx. По данным полученной таблицы была построена гистограмма, отражающая наиболее "популярное" место начала посл-ти Шайна-Дальгарно в геноме Pyrobaculum oguniense относительно начала кодирующего участка (рис. 4).
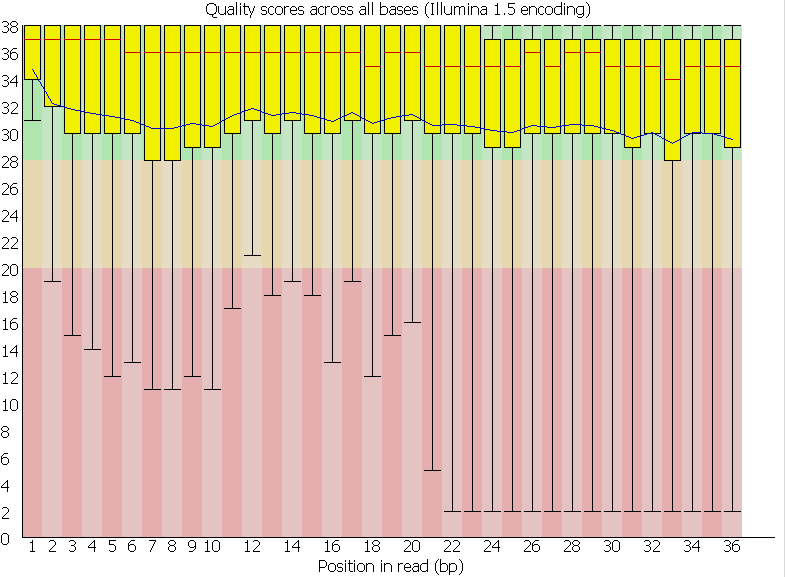
На гистограмме (Рис. 4) видно, что чаще всего мотив Шайна-Дальгарно начинается в -11 позиции от старта трансляции, однако по гистограмме видно, что эта ситуация наблюдается не в подавляющем большинстве случаев. Таким образом, либо не все последовательности Шайна-Дальгарно были найдены, либо в принципе выбранной архее свойственны иные механизмы инициации трансляции без участия ШД. В пользу этого говорят данные статьи по родственному организму Pyrobaculum aerophilum[2]. В ней говорится о том, что в мРНК этого организма отсутствует 5'-UTR, в связи с чем эта архея, а также, вероятно, и все кренархеоты (к которым относится и рассматриваемая мной Pyrobaculum oguniense) используют другие механизмы инициации без участия рибосом-связывающего сайта. Ссылки: [1] - The 3'-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites, Shine J, Dalgarno L., 1974). [2] - Genome sequence of the hyperthermophilic crenarchaeon Pyrobaculum aerophilum, Fitz-Gibbon ST, Ladner H. et al., 2002). [3] - Complete genome sequence of Pyrobaculum oguniense, David L. Bernick, Kevin Karplus et al., 2012). Задание 3. Поиск сайтов связывания транскрипционного фактора в данном участке хромосомы Был взят файл chipseq_chunk48.fastq. C помощью команды fastqc chipseq_chunk48.fastq было проверено его качество. На рис. 5. показано качество прочтения каждого нуклеотида и видно, что в целом чтение неплохое, все значения попали в зеленую область, поэтому программой Trimmomatic для чистки ридов было решено не пользоваться.
Всего в файле 19291 рид, все они длины 32-36 нуклеотидов. Чтения были картированы на геном человека командой bwa mem ../hg19/GRCh37.p13.genome.fa chipseq_chunk48.fastq > chipseq_chunk48.bam. Затем выравнивания с референсным геномом были переведены в бинарный формат командой samtools view -bSo chipseq_chunk48.sam chipseq_chunk48.bam. Они были отсортированы по началу чтения в референсе командой samtools sort chipseq_chunk48.bam -T chip_temp -o chipseq_chunk48.sorted.bam. Полученный файл был проиндексирован: samtools index chipseq_chunk48.sorted.bam, информация о количестве картированных чтений была записана в файл chipseq_chunk48.idxstats командой samtools idxstats chipseq_chunk48.sorted.bam > chipseq_chunk48.idxstats. Наконец, команда samtools idxstats chipseq_chunk48.sorted.bam > chipseq_chunk48.idxstats вывела на экран количество всех картированных чтений. Результат приведен на рис. 6; видно, что в итоге откартирован 19291 рид, то есть все исходные.
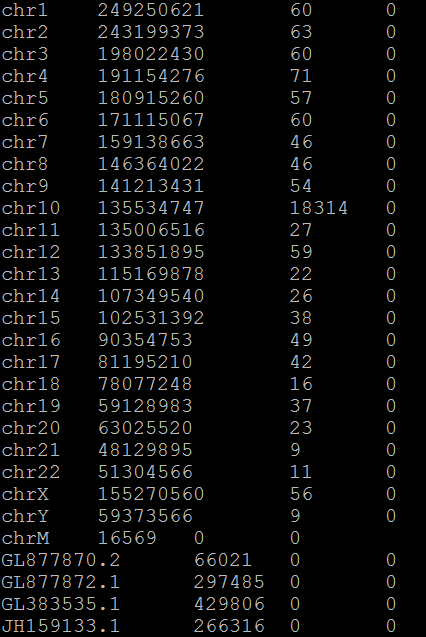
В файле chipseq_chunk48.idxstats хранится информация о том, сколько картированных ридов приходится на каждую хромосому. Его фрагмент приведен на рис. 7.
Судя по рис. 7, основная часть ридов - 18314 штук (почти 95%) - откартировалась на 10-ю человеческую хромосому. Затем был проведен peak calling с помощью программы MACS. Сначала она была запущена командой macs2 callpeak -t chipseq_chunk48.sorted.bam, однако в выдаче было сказано, что парных пиков нет, поэтому использовалась команда
macs2 callpeak -t chipseq_chunk48.sorted.bam -n chunk48 --nomodel (параметр -n задает название эксперимента).
С ее помощью были получены три файла: chunk48_peaks.narrowPeak, chunk48_peaks.xls и chunk48_summits.bed.
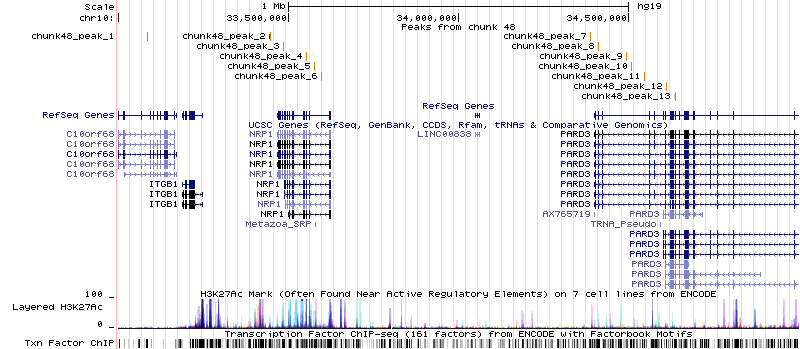
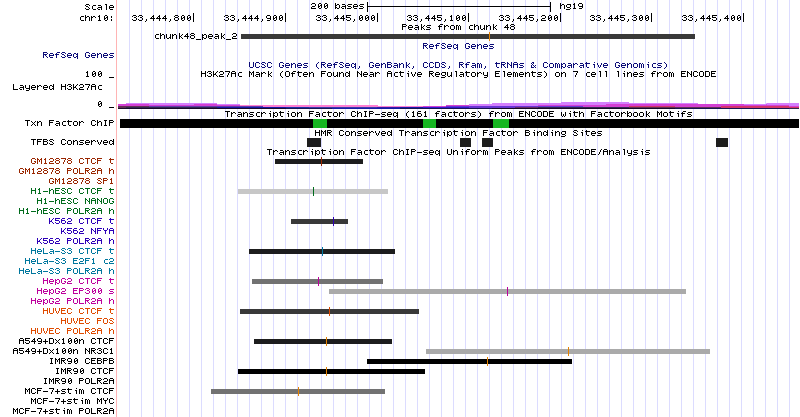
Всего было найдено 13 пиков. Их визуализация на сервере UCSC Genome Browser представлена на рис. 8. Заметно, что они группируются друг с другом: пики 2 - 6 находятся примерно в одном месте, как и последние 7 (в другом).
Самым крупным пиком является второй (ширина 496 пн), а его -log10pvalue и -log10qvalue равны 87.41162 и 77.93585 соответственно. Помимо этого он имеет fold enrichment 15.49(этот параметр показывает кратность превышения случайного распределения Пуассона). Это самые высокие показатели среди остальных пиков, что означает наибольшую достоверность (т.к. наименьшую из представленных степеней p-value). В увеличенном масштабе он представлен на рис. 9.
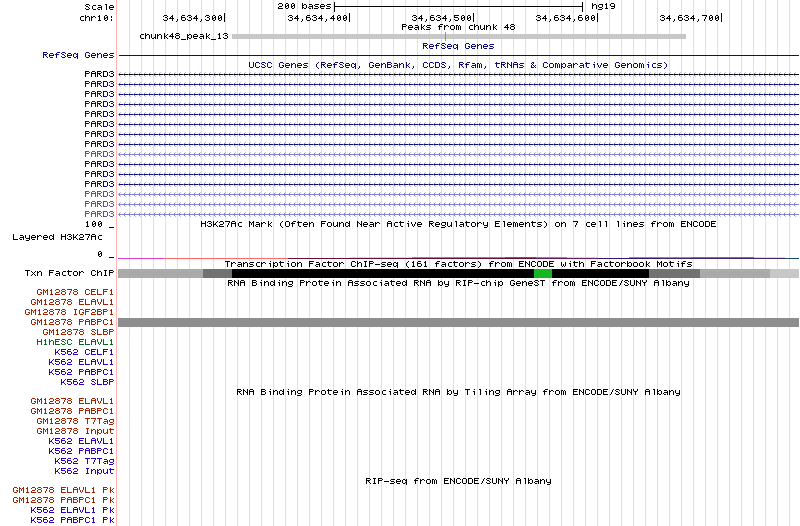
На рис. 9 показаны также данные по другим известным транскрипционным факторам. Видно, что второй пик (то есть предполагаемое место посадки изучаемого транскрипционного фактора) колокализован с несколькими из приведенных факторов. Также был подробнее рассмотрен 13-ый пик (рис. 10). Он расположен в интервале с 34,634,307 по 34,634,671 и имеет длину 365 пн, а его -log10p-value = 26.215, -log10q-value = 19.414.
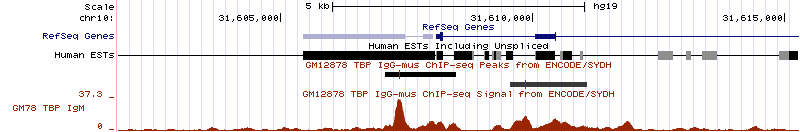
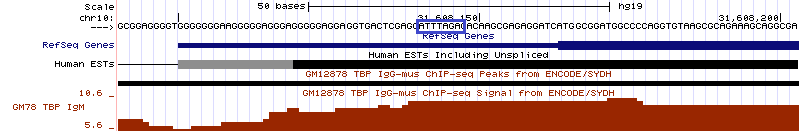
Пик попал на ген PARD3, кодирующий белок, входящий в семейство PARD. Эти белки влияют на ассиметричное деление клетки и полярность клеточного роста. Кроме того, он колокализован с PABPC1. Задание 4. Поиск сигналов TATA-box связывающего белка (TBP) в геноме человека Для работы были взяты результаты ChIP-Seq'a, использовавшиеся в предыдущем задании. ТАТА-бокс имеет консенсус TATAWAAR (W = А/Т, R = A/G). Связывание с этой последовательностью белка TBP приводит к образованию комплекса TFIID и инициации транскрипции эукариотических генов с помощью Pol II. Были проанализированы результаты ChIP-Seq для ТВР на клеточной линии GM12878 (В-лимфоциты здорового человека), в эксперименте использовались мышиные моноклональные антитела. Ниже рассмотрены разные случаи нахождения пиков сродства ТВР и нахождения ТАТА-бокса в последовательности. 1. Есть ТАТА-бокс и высокий сигнал связывания ТВР, отличимый от фона (рис. 11). Пик находился на расстоянии -16 от начала гена ZEB1, кодирующего транскрипционный фактор цинкового пальца. Его координаты: начало 31608101 (на + цепи), конец 31818742, длина гена - 210642 пн.
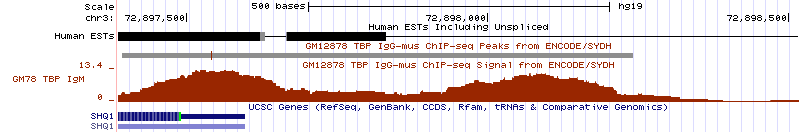
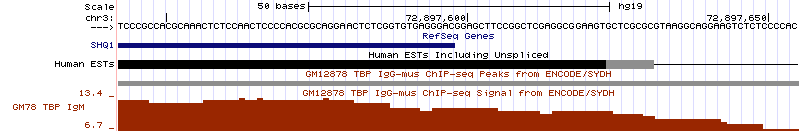
2. В 3-ей хромосоме было найдено место, где присутствуют достоверные пики для ТВР, однако в последовательности нет ТАТА-бокса (рис. 12), зато наблюдается большое количество С. Пик находится в промоторной области белка SHQ1, фактора сборки рибонуклеопротеина. Координаты гена: 72798428-72897598, длина 99171 пн, находится на - цепи.
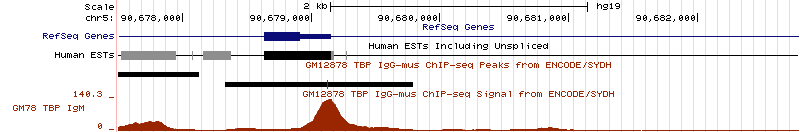
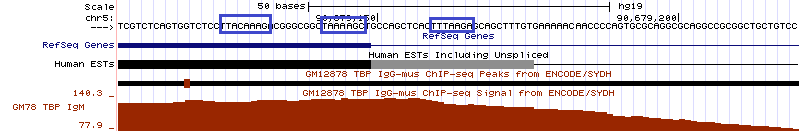
3. В 5-ой хромосоме перед геном ARRDC3. Этот ген кодирует белок,содержащий аррестиновый домен (аррестины - белки, участвующие в регуляции передачи сигнала от G-белоксвязывающих рецепторов). Координаты гена: 90,664,541-90,679,149, длина 14609 пн, находится на - цепи.
На рис. 13 приведен скриншот, на котором показано, что пик ярко выражен и существенно отличается от фона, и сайтов посадки TBP можно предположить несколько (вероятнее всего, сайт находится на позиции -11 относительно начала гена).
Таким образом, в целом, можно заметить, что ТАТА-бокс не всегда требуется для инициации транскрипции эукариот и архей, не все гены имеют такой сигнал в промоторной области. Надо отметить, что найти ТАТА-мотивы у существующих пиков сайта связывания TBP было достаточно нетривиальной задачей. Помимо этого, многие гены транскрибируются независимо от работы этого белка. | |||
| © Alexandra Boyko, 2014. Faculty of Bioengineering and Bioinformatics, MSU. | |||