Построение выравнивания последовательности из структуры ID: 1lmp и предложенного белка (в моем случае LYS_BPPS1). Выравнивание строили в программе ClustalW и сохранили в формате PIR.
Модификация файла выравнивания:
Последовательность в файле выравнивания переименовали как в примере:Было Стало >P1;uniprot|P37712|LYSC_CAMDR >P1;seq >P1;1LMP__|PDBID|CHAIN|SEQUENCE >P1;1lmp После имени последовательности моделируемого белка добавили строчку:
sequence:XXXXX::::::: 0.00: 0.00
эта строчка описывает входные параметры последовательности для modeller.
После имени последовательности белка-образца добавили:structureX:1lmp_now.ent:1 :A: 132 :A:undefined:undefined:-1.00:-1.00
эта строчка описывает, какой файл содержит структуру белка с этой последовательностью, номера первой и последней аминокислот в структуре, идентификатор цепи и т.д.
В конце каждой последовательности добавили символы/.
Символ "/" означает конец цепи белка. Точка указывает на то, что имеется один лиганд (если бы было два лиганда стояли бы две точки). В итоге получился файл aligned.pir.Модификация файла со структурой: удалили всю воду из структуры (в текстовом редакторе). Всем атомам лиганда присвоили один и тот же номер "остатка" (MODELLER считает, что один лиганд = один остаток) и модифицировали имена атомов каждого остатка, добавив в конец буквы A, B, C. Смысл операции в том, что атомы остатка 130 имели индекс А, атомы остатка 131 имели индекс В и т.д. . После модификации имен атомов изменили номера остатков на 130.
Пример:
сохранили в файле 1lmp_now.entБыло Стало HETATM 1014 O7 NAG 130 HETATM 1014 O7A NAG 130 HETATM 1015 C1 NAG 131 HETATM 1015 C1B NAG 130 Создание управляющего скрипта. Нам представлена следующая заготовка:
from modeller.automodel import * сlass mymodel(automodel): def special_restraints(self, aln): rsr = self.restraints for ids in (('OD1:98:A', 'O6A:131:A'), ('N:65:A', 'O7B:132:A'), ('OD2:73:A', 'O1C:133:A')): atoms = [self.atoms[i] for i in ids] rsr.add(forms.upper_bound(group=physical.upper_distance, feature=features.distance(*atoms), mean=3.5, stdev=0.1)) env = environ() env.io.hetatm = True a = mymodel(env, alnfile='test1.ali', knowns=('1lmp'), sequence='seq') a.starting_model = 1 a.ending_model = 5 a.make()В скрипте указано:
что нужно использовать стандартные валентные углы в полипептидной цепи (строчка 4)
что дополнительно нужно сохранять взаимное расположение определенных пар атомов (3.5 ангстрема);
В данном случае трех атомов белка, образующих водородные связи с тремя атомами лиганда - строчки 5-7 с ID пар атомов; параметры взаимного расположения атомов пары описаны в строчке 9-10. 3 точки могут однозначно расположить сложную структуру в пространстве, поэтому мы выбираем водородные связи как источник данных точек.
что ковалентные связи в гетероатомах нужно вычислять по расстояниям между атомами (так же, как это делает Rasmol), строчка 12
что имя файла с выравниванием и имена последовательностей образца и моделируемого белка, строчка 13 (а имя файла со структурой содержится в выравнивании)
что число и номера моделей, которые нужно построить (в данном примере 5 моделей), строки 14-15
что пора строить модель строчка 16
В скрипте необходимо отредактировать строчки, в которых указаны какие водородные связи белка с лигандом должны быть в будущей модели.
Для этого сперва определили номера остатков и имена нужных атомов по тому, какие водородные связи имеются в образце (критерий водородной связи: расстояние менее 3.5 ангстрем между азотом или кислородом белка с подходящими атомами лиганда)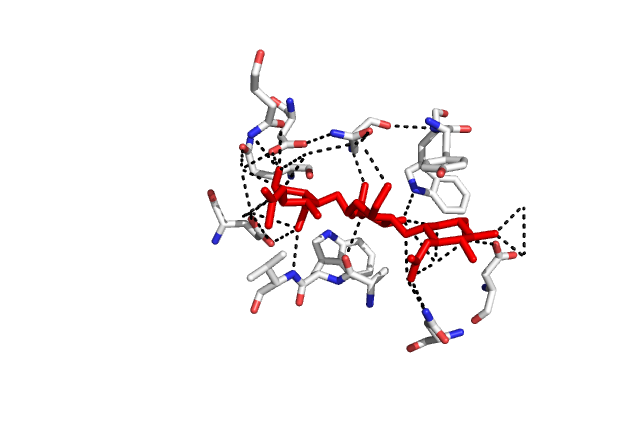 А затем по уже построенному выравниванию образца с нашим белком, так как в моделируемом белке число остатков не совпадает с числом остатков в
белке-образце и номера "остатков" лиганда изменились.
А затем по уже построенному выравниванию образца с нашим белком, так как в моделируемом белке число остатков не совпадает с числом остатков в
белке-образце и номера "остатков" лиганда изменились.
В итоге имеем скрипт lys_bpps1.py.Запускаем исполнение скрипта командой
mod9v7 lys_bpps1.py &
Рассмотрим полученные модели.
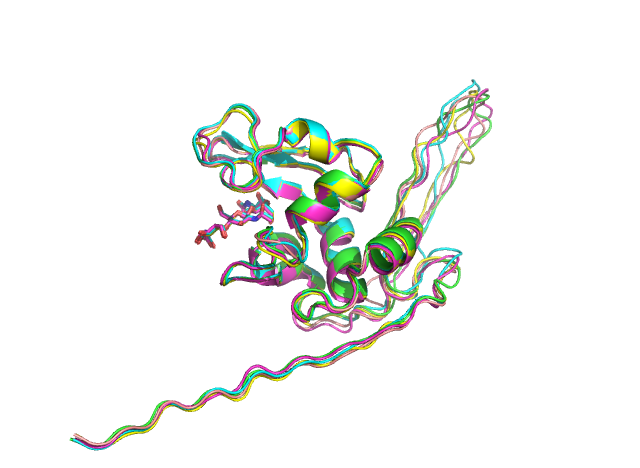
Модели очень похожи, наибольшие расхождения наблюдаются в петлях и N-конце, который тоже никак не структурирован (представлен прямой цепью, так как в структуре образца его не было).Проверим качество моделей и выберем лучшую. Инструменты для оценки качества структуры можно найти в веб интерфейсе WHATIF. Достаточно 2-3 инструментов.
1 Model Structure Z-scores, positive is better than average: Ramachandran Z-score : -0.810 RMS Z-scores, should be close to 1.0: for bond lengths : 0.988 for bond angles : 1.312 Improper dihedral : 1.104 2 Model Structure Z-scores, positive is better than average: Ramachandran Z-score : -1.137 RMS Z-scores, should be close to 1.0: for bond lengths : 0.975 for bond angles : 1.295 Improper dihedral : 1.121 3 Model Structure Z-scores, positive is better than average: Ramachandran Z-score : -1.264 RMS Z-scores, should be close to 1.0: for bond lengths : 0.985 for bond angles : 1.272 Improper dihedral : 1.095 4 Model Structure Z-scores, positive is better than average: Ramachandran Z-score : -1.288 RMS Z-scores, should be close to 1.0: for bond lengths : 0.985 for bond angles : 1.338 Improper dihedral : 1.052 5 Model Structure Z-scores, positive is better than average: Ramachandran Z-score : -1.267 RMS Z-scores, should be close to 1.0: for bond lengths : 0.999 for bond angles : 1.319 Improper dihedral : 1.046
Лучшая модель по z-score Рамачандрана: 1
По Bond lengths: 5
По Bond angles: 3
По Improper dihedral: 5
Кажется, 5 модель лучше остальных.