Сравнение матриц аминокислотных замен
В настоящее время используются разные алгоритмы и методы выравнивания аминокислотных последовательностей. Показателем качества выравнивания является его вес. Чем выше вес, тем больше сходство между последовательностями и выше качество выравнивания. Чтобы вычислить вес всего выравнивания необходимо определить частный вес каждой пары замен при выравнивании последовательностей. Аминокислоты с близкими биохимическими свойствами характеризуются большей вероятностью парных замен. Для того чтобы учесть неравную вероятность замен были разработаны специальные матрицы, которые получили название матриц замен. Наиболее широко используемыми матрицами на сегодняшний день являются матрицы BLOSUM и PHAT. Обе матрицы созданы на основе базы данных BLOCKS.
Матрица BLOSUM была предложена Стивеном и Джорджей Хеникофф в 1992 году. Она используется для выравнивания последовательностей цитоплазматических белков. Каждая матрица соответствует специфическому проценту кластеризации. Например, для матрицы BLOSUM62 процент кластеризации равен 62%. Это значит, что все последовательности выравнивания, процент идентичности которых равен или больше 62%, объединяют в единый кластер. Тогда для этих последовательностей рассчитывается определенный коэффициент замен аминокислот. Это позволяет избежать ошибку при подсчете частот пар выравнивания очень близких последовательностей. Очевидно, что матрицы с меньшим порогом кластеризации предназначены для выравнивания более удаленных друг от друга последовательностей.
Для мембранных белков, которые достаточно сильно отличаютя от цитоплазматических, разработана специальная матрица замен PHAT.
На основе таблицы частот встречаемости всех пар аминокислот мною была построена матрица BLOSUM62. Саму матрицу и расчеты, использованные при построении можно пронаблюдать в следующей таблице: evstafyeva_pr6.xlsx. Для сравнения велечин весов замен аминокислот разных матриц был использован серин. Результаты приведены в табл.1.
| G | P | C | S | T | N | Q | D | E | H | R | K | A | M | I | L | V | F | W | Y | |
| Gly | Pro | Cys | Ser | Thr | Asn | Gln | Asp | Glu | His | Arg | Lys | Ala | Met | Ile | Leu | Val | Phe | Trp | Tyr | |
| Восстановленная матрица | -1 | -1 | 0 | 4 | 2 | 0 | -1 | -1 | -1 | -1 | -1 | -1 | 1 | -2 | -3 | -3 | -2 | -2 | -2 | -2 |
| BLOSUM62 | 0 | -1 | -1 | 4 | 1 | 1 | 0 | 0 | 0 | -1 | -1 | 0 | 1 | -1 | -2 | -2 | -2 | -2 | -3 | -2 |
| PHAT_T75_B73 | 1 | -3 | 1 | 6 | 1 | 1 | -1 | -4 | -3 | -2 | -6 | -5 | 2 | -2 | -2 | -2 | -2 | -2 | -5 | -2 |
Табл.1. Сравнение матриц замен для серина (Ser)
Cравнение величин для замены серина
- В востановленной матрице и в матрице BLOSUM62 вес замены серина на самого себя равен 4, в отличие от матрицы PHAT_T75_B73, в которой этот вес равен 6.
- Серин - это гидрофильная незаряженная аминокислота. Близкими ей по химическим свойствам являются треонин(Thr, t), цистеин(Cys, C), аспарагин(Asn, N) и глутамин(Gln, Q). Значения для треонина и аспарагина в матрицах BLOSUM62 и PHAT одинаковы, что нельзя сказать о восстановленной матрице, в которой эти значения отличаются на несколько единиц. Что касается цистеина, то величина замены серина на него для разных матриц различна. Для восстановленной матрицы это значение равно 0, для BLOSUM62 -1, а для PHAT 1. Для глутамина вес замены равен -1 в восстановленной матрице и в PHAT, а в BLOSUM62 это величина равно нулю.
- Изучая таб.1, можно заметить, что вес замен серина на аминокислоты других функциональных групп почти всегда принимает отрицательно значение, кроме аланина и глицина. Рассмотрим фенилаланин, это гидрофобная ароматическая аминокислота. Значение замены для трех матриц равно -2. Это значит, что вероятность замены гидрофильной аминокислоты на гидрофобную довольно низкая. Рассмотрим лизин, это гидрофильная положительно заряженная аминокислота, в отличие от незаряженного серина. Для восстановленной матрицы вес замены равен -1, для BLOSUM62 - 0, а для PHAT 5. Это означает, что замена в мембранных белках происходит довольно часто.
Сравнение выравниваний, полученных для коротких мутантов вручную и построенных классическими алгоритмами Нидлмана-Вунша и Смита-Ватермана
Для нахождения оптимального выравнивания существуют специальные алгоритмы. Одними из них являются алгоритм Нидлмана — Вунша и алгоритм Смита-Ватермана.
Алгоритм Нидлмана — Вунша был предложен в 1970 году Солом Нидлманом и Кристианом Вуншем. Этот алгоритм реализуется с помощью программы needle. Needle выравнивает всю последовательность, то есть по всей ее длине. Алгоритм Смита-Ватермана реализуется с помощью программы water. Эта программа выравнивает не всю последовательность, как это происходит с needle, а отдельный участок, оставшееся отбрасывается.
Параметры, используемые каждым алгоритмом:
- -gapopened - штраф за один гэп. Равен 10-и по умолчанию.
- -gapextended - штраф за несколько гэпов, стоящих рядом. Равен 0,5 по умолчанию.
В трех последующих таблицах представлены результаты сравнения белка URHG2_BACSU с искуственно полученными последовательностями мутантов этого белка, состоящих из 20-и аминокислот. Эти мутанты были получены с помощью специального скрипта, моделирующего эволюцию последовательности. Подробнне об этих искусственно созданных мутантах можно узнать в документе: evstafyeva_pr5.docx.
| Выравнивание | Identity | Similarity | Score | |
| Ручное выравнивание |  |
7/20*100%=35% | 10/20*10%=50% | 22 |
| Needle | 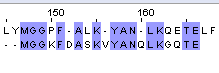 |
13/376 ( 3.5%) | 14/376 ( 3.7%) | 35.0 |
| Water | |
13/20 (65.0%) | 14/20 (70.0%) | 35.0 |
Таб.2. Выравнивание белка с мутантом №1_1
| Выравнивание | Identity | Similarity | Score | |
| Ручное выравнивание | 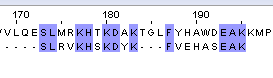 |
11/20*100%=55% | 12/20*100%=60% | 48 |
| Needle | 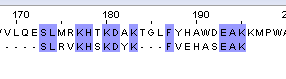 |
11/373 ( 2.9%) | 12/373 ( 3.2%) | 35.0 |
| Water | |
11/23 (47.8%) | 12/23 (52.2%) | 35.0 |
Таб.2. Выравнивание белка с мутантом №2_1
| Выравнивание | Identity | Similarity | Score | |
| Ручное выравнивание | 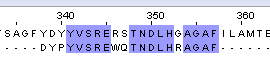 |
14/20*100% = 70% | 15/20*100%=75% | 57 |
| Needle | 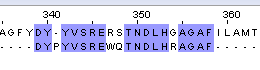 |
16/374 ( 4.3%) | 16/374 ( 4.3%) | 72.0 |
| Water | |
16/20 (80.0%) | 16/20 (80.0%) | 72.0 |
Таб.2. Выравнивание белка с мутантом №3_1