Восстановление предкового состояния доменной архитектуры
Выбор объекта исследования и построение выравнивания
Выбрать домен предлагалось несколькими способами. Сначала я стала выбрала семейство Peptidase_M48, к которому принадлежит белок, выданный мне на первом курсе, но оказалось, что оно не подходит по рекомендованным критериям (последовательностей в нём было больше 10000 и видов больше 1200). Тогда мой выбор остановился на домене D-aminoacyl_C (AC = PF07908, ID = D-aminoacyl_C). Функция этого семейства белков заключается в гидролизе большого числа N-ацильных произодных D-аминокислот. Для данного домена было найдено 23 архитектуры, соответствующих 859 последовательностям. Выравнивание всех последовательностей, скаченных с помощью Fetch sequences по идентификатору PF07908, можно скачать: jvp-формат,fasta-формат. (окрашивание ClustalX, порог консервативности 12%). К выравниванию была добавлена 3D структура последовательности Q9AGH8 (1M7J).
Далее, с помощью скриптов, выложенных на сайте, и базы данных Uniprot, была получена таблица с указанием наличия доменов, таксономии и выборкой белков для работы. Для дальнейшей работы был выбран таксон Bacteria и две архитектуры - Amidohydro_3, D-aminoacyl_C" и "Amidohydro_5, D-aminoacyl_C" (указано на листе 'Selected'). Для каждой архитектуры было отобрано примерно 20 белков и построено по ним выравнивание: jvp-формат, fasta-формат (окраска ClustalX, порог консервативности 10%).
Построение филогенетического дерева последовательностей домена
Были введены обозначения для названия последовательностей: "Amidohydro_3, D-aminoacyl_C" - 1 и "Amidohydro_5, D-aminoacyl_C" - 2 (обе архитектуры двухдоменные, поэтому это просто порядковые номера); P - Proteobacteria, A - Actinobacteria, B - Bacteroidetes, F - Firmicutes (использовались подтаксоны, так как все выбранные белки относятся к одному таксону - Bacteria). После переименования было получено следующее выравнивание: jvp-формат, fasta-формат. Для построения дерева по этому выравниванию был выбран метод Neighbor-Joining using % identity cо 100 репликами Bootstrap (рис. 1). также была сохранена скобочная формула для дерева в формате Newick.
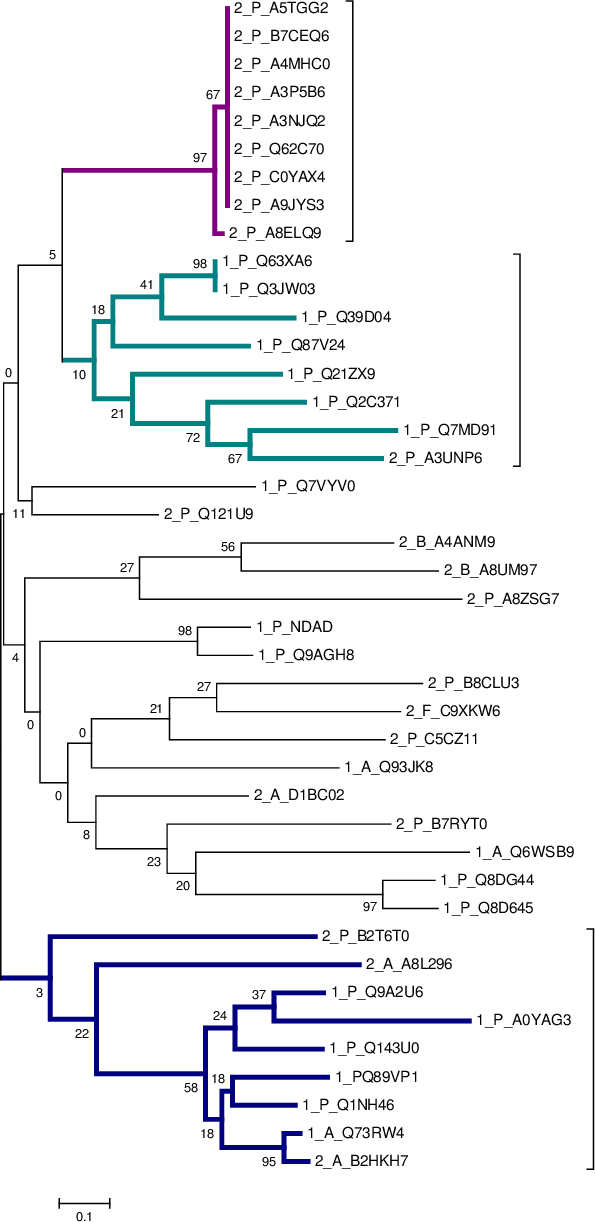
Рисунок 1. Филогенетическое дерево, построенное в программе MEGA методом процентного сходства с соседями cо 100 репликами бустрэп.
На рисунке хорошо видно, что отделилась одна клада (выделена фиолетовым) - подтаксон Proteobacteria с архитектурой 2. Вторая довольно хорошая клада (выделена бирюзовым) состоит из подтаксона Proteobacteria с архитектурой 1 и одной последовательностью архитектуры 2. Остальные клады не имеют точного разделения на подтаксоны, но довольно хорошо разделяются по архитектурам. В кладе, выделенной синим цветом, над архитектурой 2 начинает превалировать архитектура 1 (хотя изначально в ней основной была 2), что позволяет предположить, что в ходе происходит замена одной архитектуры на другую.
Построение профиля подсемейства и охарактеризование качества его работы
Был построен профиль для хорошей клады подтаксона Proteobacteria с архитектурой 2 (на рис.1 выделена фиолетовым). Профиль был откалиброван и применен к fasta-файлу со всеми найденными доменами D-aminoacyl_C. На выходе был получен следующий файл: результат. На его основе была построенаа ROC-кривая, показанная на рисунке 2.
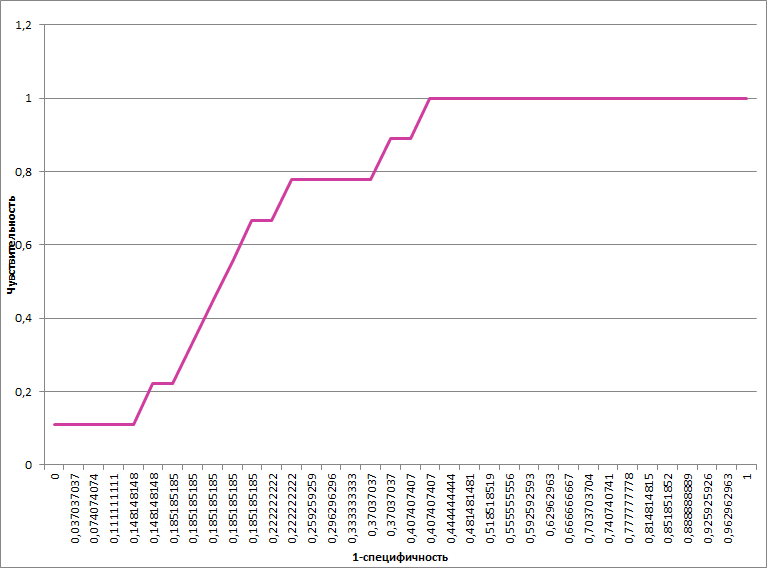
Рисунок 2. ROC-кривая
Порогом для кривой был выбран уровень e-value в 1.6, при таком пороге результаты применения профиля следующие:
Таблица 1. Результаты применения профиля
| принадлежит | не принадлежит | сумма | |
|---|---|---|---|
| выше порога | 9 | 0 | 9 |
| ниже порога | 13 | 14 | 27 |
| сумма | 22 | 14 | 36 |
При данном уровне e-value данный профиль можно использовать для определения подсемейства.
Дата последнего изменения: 15.09.2013
