Множественные выравнивания. Pfam.
Множественные выравнивания
При помощи сервиса BLAST мною была собрана выборка из 8 белков, гомологичных белку β-порфираназе А бактерии Zobellia galactanivorans (включая сам белок).
Скачать выборку можно по по этой ссылке. Мною было построено множественное выравнивание отобранных последовательностей при помощи программы muscle на сервере kodomo. Введеная мною команда приведена ниже:
muscle -in aligment_1.txt -out aligment_mus.fa -diags -maxiters 32 -maxhours 0.5 -log lof_alig_1.txt
При выполнение команды информация о работе программы была записана в файл, который можно скачать по этой ссылке.
Увидеть построенное выравнивание
можно на рис. 1. Скачать его отдельно можно по этой ссылке.

Рис. 1. Множественное выравнивание с помощью программы muscle.
Далее мною были построены множественные выравнивания с помощью нескольких программ( в случае mafft - для разных опций), затем была произведено их улучшение с помощью алгоритма оптимизации muscle, произведено сравненение множественных выравниваний до и после выравнивания. Все программы одинаково хорошо нашли участки схожести последовательностей, разлчия в середине были крайне незначительны, на концах - чуть более существенные.(таблица сравнения)
Таблица 1. Сравнение работы различных программ для построения множественного выравнивания. Спасибо Андрею Владимировичу Алексеевскому за объяснение значения различных опций mafft.
| Программа | Изменение алгоритмом оптимизации (refimnent), реализованным в muscle | Ссылка на проект с приведенными множественными выравниваниями до и после алгоритма оптимизациии |
| mafft( без опций) | Различия в начале, небольшие в середине и в конце. Основные, хорошо выравненные участки не меняются | скачать |
| mafft --globalpair(globalpair - строит глобальное выравнивание) | незначительное изменения, затронуты только очень плохо выравнивающиеся участки | скачать |
| mafft --localpair(строит (почти) глобальное выраванивание, не выравниваются только концевые фрагменты, если они не похожи) | Изменения в плохо выравнивающихся участках. | скачать |
| mafft --genafpair(строит выравнивание на участках, где оно детектируется, а остальные участки не выравниваются) | небольшие изменения в неконсервативных колонках | скачать |
| ClustalOmega | большие изменения, но в плохо выравнивающихся участках | скачать |
| ClustalW | большие изменения, выравнивание улучшилось | скачать |
| T-coffee | Незначительные изменения в неконсервативных участках | скачать |
Для сравнения было выбрано выравнивание, построенное Tcoffee. C ним будет сравниваться выравнивание, построенное выше с помощью muscle.
Для сравнения выравниваний был использована команда:
muscle -profile -in1 aln_tcoffee.fa -in2 aligment_mus.fa -out both_alig.fa
Получившееся выравнивание можно увидеть на рисунке 2. Стоит отметить, что выравнивания очень схожи и отличаются лишь на полностью несхожих участках. Их схожесть
подтверждают и практически идентичные деревья сходства, построенные в Jalview по этим выравниваниям(рис. 3 и рис. 4). Участки свопадения были отмечены "+", несовпадения - "-". Для облегчения работы применялся скрипт, который
можно скачать по этой ссылке. Для корректной работы скрипта последовательности в первом и втором выравнивание должны быть расположены в одном порядке, на вход принимается выравнивание выравниваний. На выход - файл, содержащий последовтельность из плюсов и минусов.

Рис. 2. Выравнивание множественных выравниваний, построенных с помощью программы muscle и T-coffee.
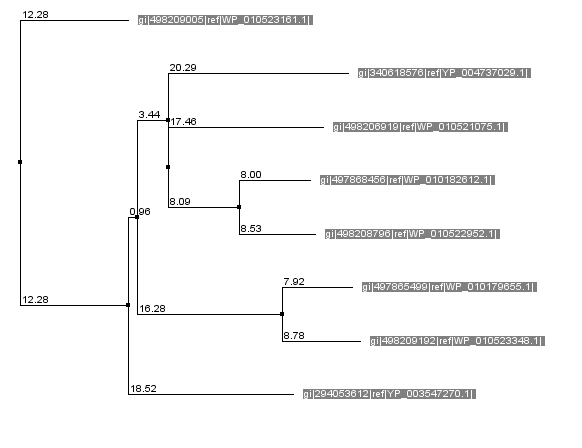
Рис. 3. Дерево сходства, построенное по множественному выравниванию, полученному с помощью T-coffee.
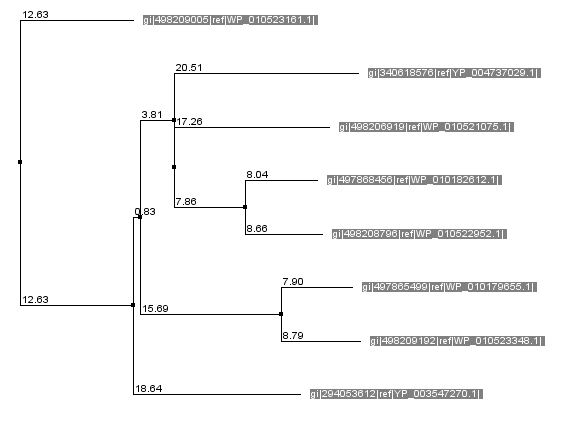
Рис. 4. Дерево сходства, построенное по множественному выравниванию, полученному с помощью muscle.
После этого я с помощью сайта Pfam посмотрел, домены каких семейств, встречаются в белке β-порфираназе А. Выдачу Pfam можно увидеть на рис. 5. В программе используются Скрытые Марковские модели(HMM), строка #HMM содержит консенсусную последовательность, полученную из HMM, большими буквами показаны наиболее консервативные аминокислоты. #Match показывает соответствие между HMM и нашей последовательностью, #PP - содержит число, характеризующую уверенность в том, что выравнивание в определенной позиции верно. 0 - от 0 до 5%, 1 - 5-15% и т.д. * - от 95% до 100%. #SEQ - наша последовательность, выравненная относительно домена.
Согласно выдаче Pfam в моём белке есть один консервативный домен семейства Glycoside hydrolase family 16. Белки этого семейства представляют собой широкую группу белков, гидролизирующих гликозидные связи между двумя и более углеводами или между углеводной и неуглеводной частями[1]. Семья принадлежит к клану "Concanavalin-like lectin/glucanase superfamily", включающему 16 семейств и общее число доменов равное 34755. Клан был создан А. Байтманом[2].
На рис. 6 приведена схема, отображающая положение Glycoside hydrolase family 16 в клане.
С помощью сервиса Pfam также можно построить круговые диаграммы, отображающие наличие белков семейства в разных таксонах. Распределение по количеству секвенированных последовательностей показано на рис. 7 . Распределение по количеству организмов на рис. 8 . Видно, что хотя число последовательностей белков этого семейства, принадлежащих эукариотам больше, это вызвано скорее многократным секвенированием геномов эукариотных организмов, нежели их количеством. Стоит отметить, что среди бактерий очень обширный состав видов, имеющих гены белков данного семейства, среди эукариот таких организмов меньше - среди Fungi и Metazoa( особенно интересно, что у комаров Anopheles, переносчиков малярийных плазмодиев, также встречается этот белок.), к примеру.(Хотя в случае Metazoa в некоторых случаях, на мой взгляд, имеются в виду белки бактериальных симбионтов.)
Также мною было скачано seed-выравнивание белков семейства. Ссылка на файл с ним находится ниже в "Результатах".
Кроме этого с помощью сервиса PFam была построена графическая интерпретация профиля семейства Glyco_hydro_16 в Скрытой Марковской модели в Pfam(рис.9). Стоит отметить, что этот лого значительно более информативно, чем лого, потсроенное в задании 10. В лого, построенном там, отражена информация лишь об соотношении различных аминокислот в позициях и, как я понял, там не учитывалась встречаемость аминокислот в природе.
Кроме этого в Pfam logo отражена информация о возможности появления инсерций/делеций в соответсвующей позиции. За эту информацию отвечают полосы красного и розового цвета в соответствующей позиции. Ширина красной полосы характеризует возможность инсерции/делеции, а суммарная ширина красной и розовой полос отражает ожидаемое количество вставленных аминокислот.
Различие легко пояcнить на следующем примере - если в выравнивании десяти последовательностей "встречается" в пяти последовательностях по одному гэпу и в других десяти в одной последовательности есть 5 гэпов, то ожидаемое количество гэпов одинаково(соответсвенно и ширина полос), а вероятность появления гэпа в последовательности(соответсвенно и ширина красной полосы) больше в первом случае. Для примера - смотрите 41 и 55 позиции. Ожидаемое количество гэпов одинаково, но во втором случае вероятность появления гэпа вообще больше.
Результаты
Результаты всей приведенной выше работы можно скачать в виде проета JalView по этой ссылке. В состав проекта входят:
- Множественное выравнивание, полученное с помощью muscle - aligment_mus
- Множественное выравнивание, полученное с помощью tcoffee - aln_tcoffee
- Выравнивание двух предыдущих выравниваний, полученное с помощью muscle - both_alig
- Выравнивание 120 последовательностей белков семейства Glyco_hydro_16 - Glyco_hydro_16_seed
Рис. 5. Результат поиска по последовательности в Pfam.
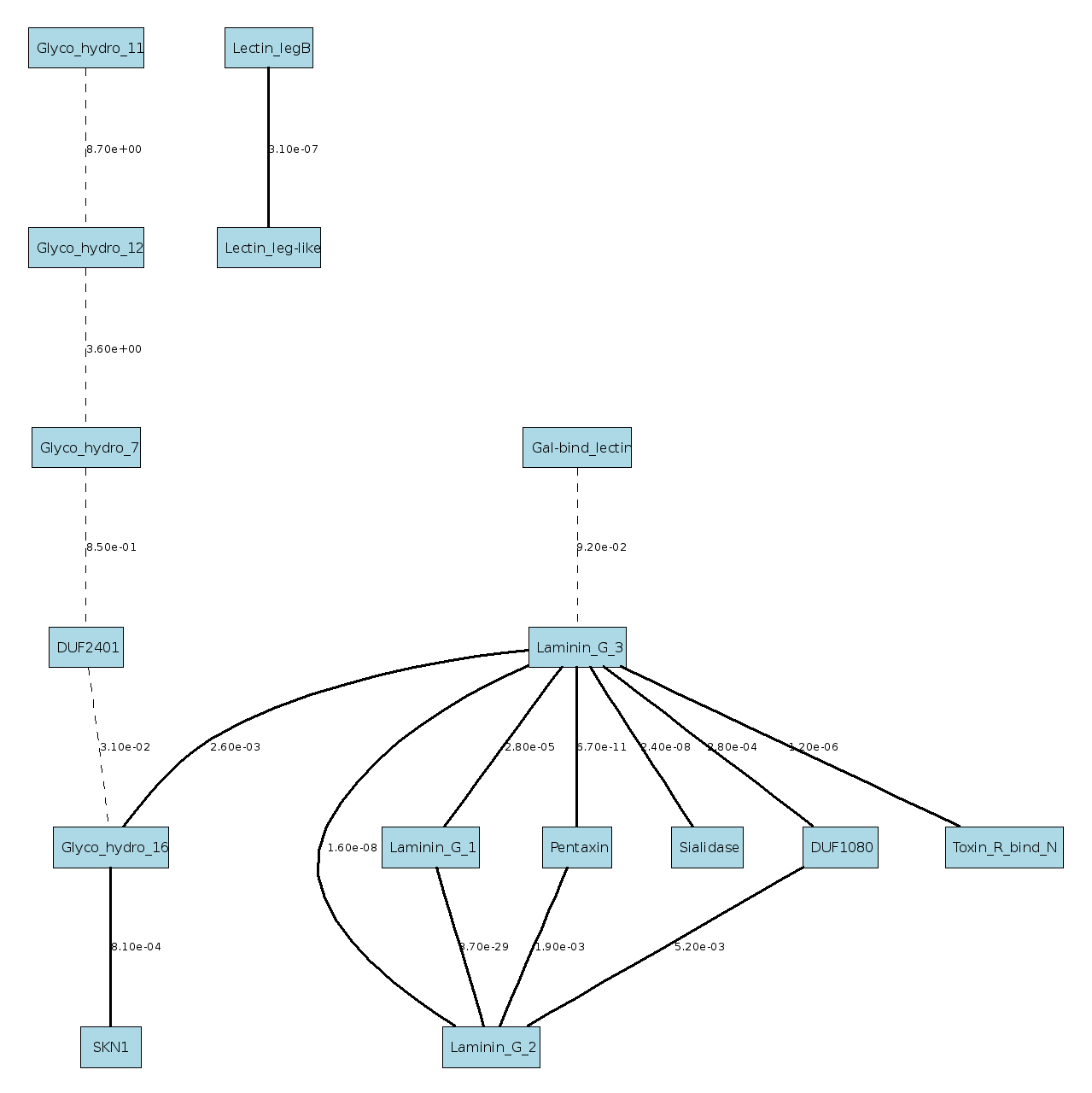
Рис. 6. Клан Concanavalin-like lectin/glucanase superfamily. Схема взята из Pfam.
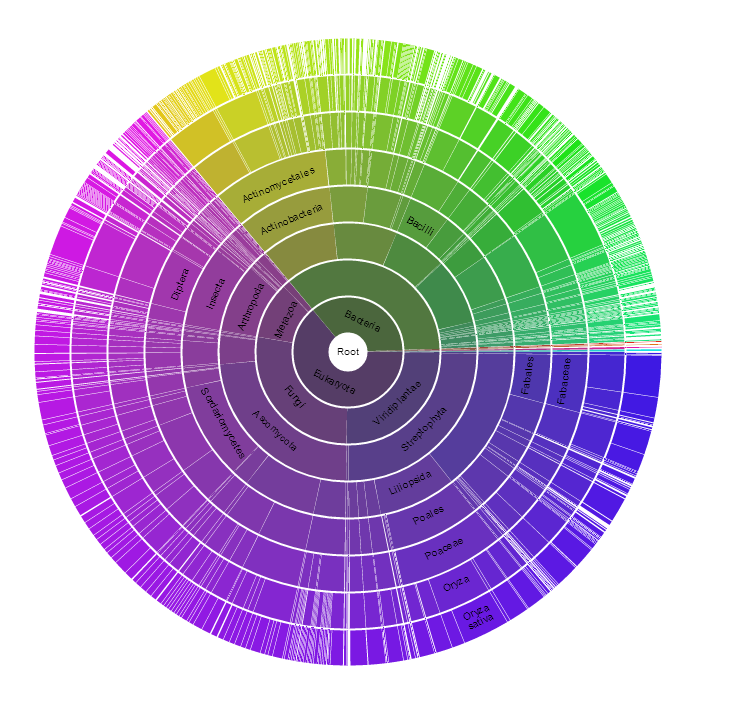
Рис. 7. Результат поиска по последовательности в Pfam.
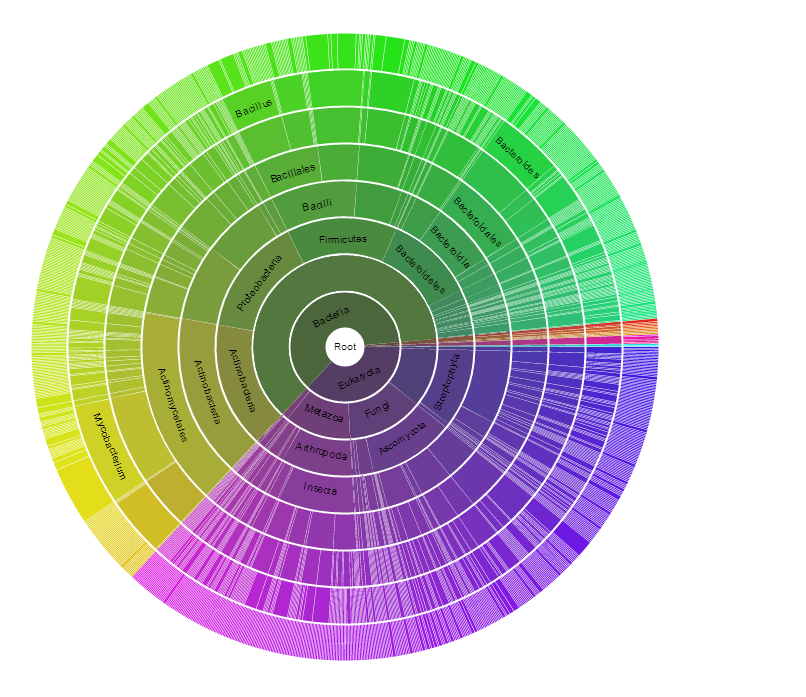
Рис. 8. Результат поиска по последовательности в Pfam.
Рис. 9. Графическая интерпретация профиля семейства Glyco_hydro_16 в Скрытой Марковской модели в Pfam.
Ссылки
