| FBB MSU site | Main page | About me | Terms |
Assembly de novo
I created a subdirectory /nfs/srv/databases/ngs/potanina.darya/pr14. This directory contains the sample of RNA sequencing of thale cress (Arabidopsis thaliana) and all prodused during making the tasks files.
Materials preparing
I downloaded a few files contain sequences of adapters from /P/y16/term3/block3/adapters/ and joined them by seqret in adapters.fa.
Reads preparation | Trimmomatic
RNA sequencing reads was cleaned of adapters by Trimmomatic installed on kodomo. The processing step ILLUMINACLIP has syntax: ILLUMINACLIP:<fastaWithAdaptersEtc>:<seed mismatches>:<palindrome clip threshold>:<simple clip threshold>.
| $ java -jar /usr/share/java/trimmomatic.jar SE -phred33 F.fastq F_without_adapters.fastq ILLUMINACLIP:adapters.fa:2:7:7 |
Output:
| Input Reads: 3869869 Surviving: 3869420 (99,99%) Dropped: 449 (0,01%) |
According the task reads cut of adapters was processed by steps SLIDINGWINDOW and MINLEN of Trimmomatic with given values SLIDINGWINDOW:5:28 (the length of window is 5 nucleotides, the min average quality is 28) and MINLEN:32 (to leave only more then 32 nucleotides length reads).
| $ java -jar /usr/share/java/trimmomatic.jar SE -phred33 F_without_adapters.fastq F_cleaned.fastq SLIDINGWINDOW:5:28 MINLEN:32 |
Output:
| Input Reads: 3869420 Surviving: 3444237 (89,01%) Dropped: 425183 (10,99%) |
Then for checking I conducted quality control of received reads by FastQC installed on kodomo.
| $ fastqc F_without_adapters.fastq $ fastqc F_cleaned.fastq |
The result you can see here:
Output files:
|
F_without_adapters_fastqc.html F_cleaned_fastqc.html |
| Per base sequence quality | |
| Figure 1. Quality of reads without adapters before cleaning. | Figure 2. Quality of reads after cleaning. |
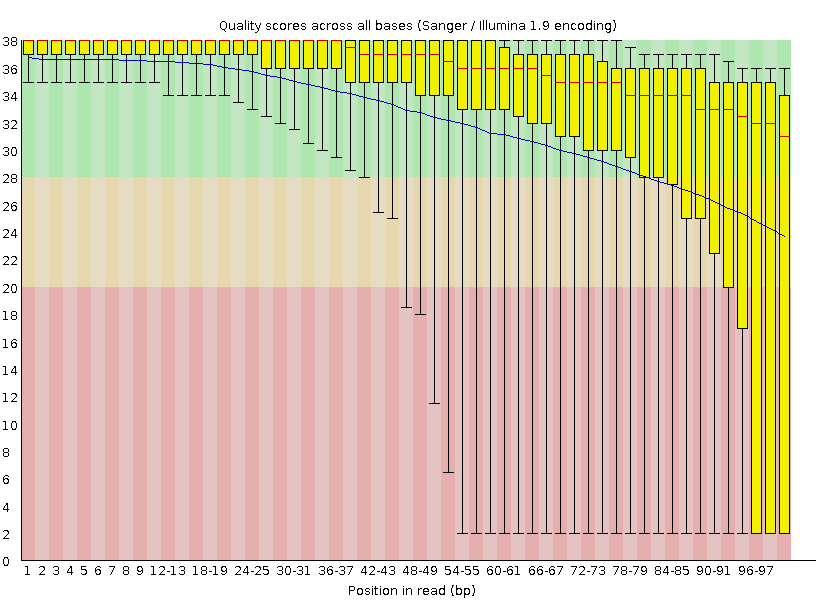 | 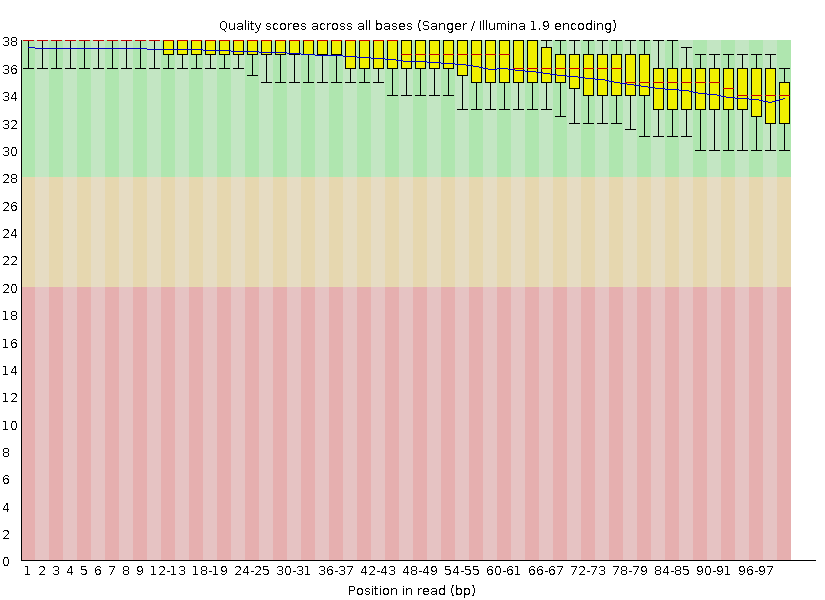 |
Outcome: the original file's size (F.fastq) is 1022153 bit or 3869869 db, after cutting of adapters (F_without_adapters.fastq): 1007715 bit or 3869420 db, after cleaning (F_cleaned.fastq): 829597 bit or 3444237 db.
Accembly | Velveth
For the genome assembly I prepaired K-mers by Velveth (k = 31). The syntax is: velveth output_directory hash_length [[-file_format][-read_type] filename]. In this case hash_length = 31, file_format = fastq, read_type = short.
| $ velveth F_velveth 31 -fastq -short F_cleaned.fastq |
Output files:
|
F_velveth/: Log Roadmaps Sequences |
Then accembly of contigs was run by velvetg.
| $ velvetg F_velveth |
Output:
| Final graph has 473340 nodes and n50 of 27, max 657, total 7126555, using 0/3444237 reads |
Output files:
|
F_velveth/: Graph LastGraph Log PreGraph Roadmaps Sequences contigs.fa stats.txt |
As we can see in output N50 = 27, the length of the longest contig = 657. Analysed 'stats.txt' I found out some data about contigs (see table 1). All values of length are measured in k-mers. To recieve real lengthes of contigs you should add (k - 1) = 30 to shown values.
The one interesting point is that arabidopsis' genome assembly differs from other organisms' genome assemblies, as I have noticed studying the work of other students. There are short contigs and naturally the number of nodes of final graph is high in arabidopsis. But the majority of students worked with other organisms received the final graph contained less nodes because of long contigs. I don't know what this differences is caused of, perhaps it is so because of the characteristics of the initial data.
Table 1.
| length (k-mers) | short1_cov (slightly different) | short1_Ocov (fully matching) |
| Three contigs have max length | ||
| 657 | 10,707763 | 10,330289 |
| 634 | 13,182965 | 10,330289 |
| 627 | 44,363636 | 10,330289 |
| The contig has max coverage | ||
| 1 | 4695117 | 4695117 |
| The contig has min coverage | ||
| * (more then one contig) | 1 | 1 |
| Average | ||
| 15,055890 | 247,369083 | 154,521475 |
| Median | ||
| 6 | 3,5 | 3,333333 |
Annotation
The longest contig
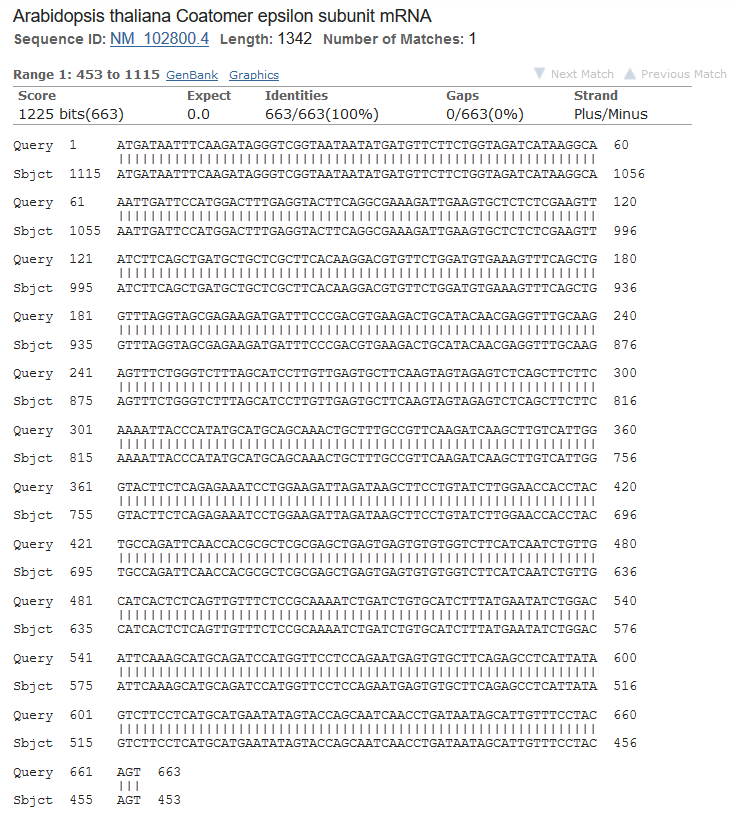
Firstly I received the longest contig (found in 'stats.txt') from 'contigs.fa'. It's name is NODE_49528_length_657_cov_10.707763 (find by comand: $ grep "length_657" contigs.fa). It was cut off by 'seqretsplit' and saved in FASTA file. Using BLAST I found the most similar sequences. I used MegaBlast, E-value = 0.0001, word size = 256, db - Nucleotide collection (nr/nt).
There were 6 findings (see fig 1). The best finding was Arabidopsis thaliana Coatomer epsilon subunit mRNA (see fig 2).
Figure 1.
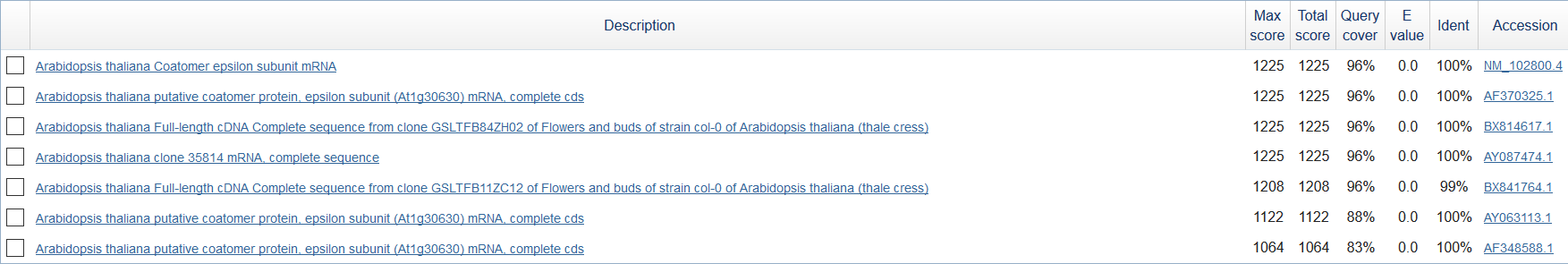
Figure 2.
The best covered contig
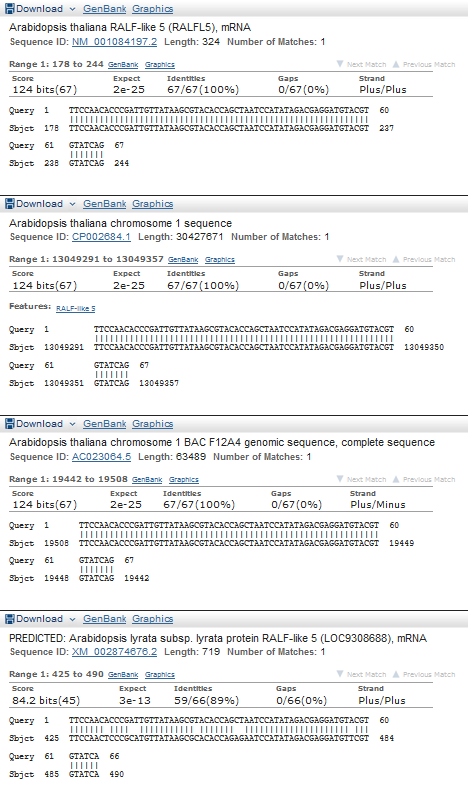
Most of the contigs with high coverage are 1-10 nucleotide length, I think it is useless to align them. So I chose contig longer than 30 which has the highest coverage in 'stats.txt' - this one: NODE_76045_length_37_cov_505.621613 (length = 37, cov = 505.621613). It was analised by MegaBlast (word size = 16). There were 4 findings (see fig 3). The best finding is Arabidopsis thaliana RALF-like 5 (RALFL5), mRNA (see fig 4).
Figure 3.
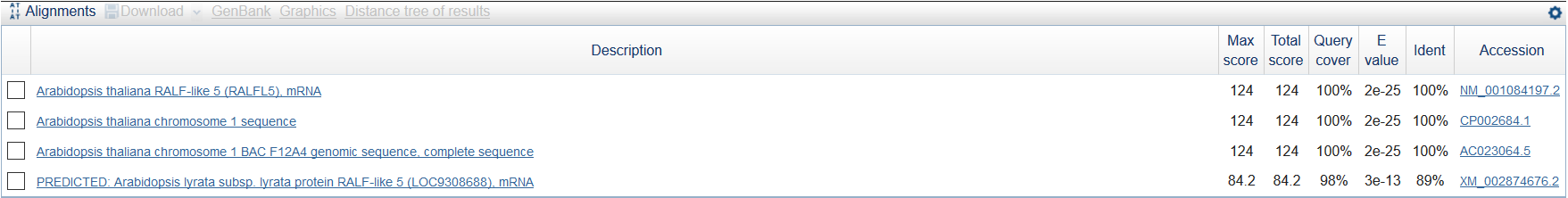
Figure 4.
The least covered contig
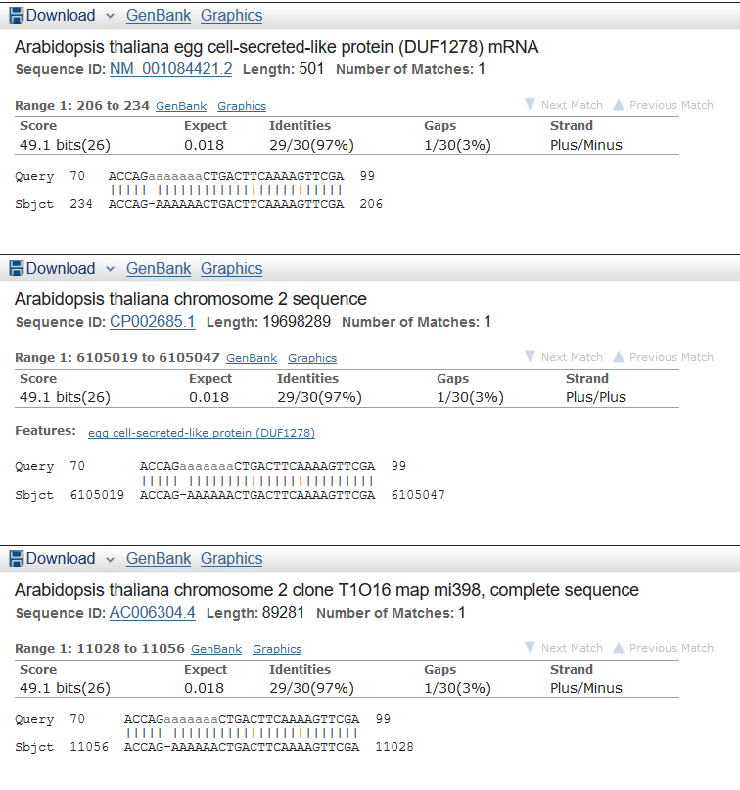
I chose the longest contig has minimum coverage in 'stats.txt' - NODE_472902_length_69_cov_1.000000 (length = 69, cov = 1.000000) and analised it by MegaBlast (word size = 16). There were 4 findings (see fig 5). Three findings have the same characteristics so I chose as the best one this: Arabidopsis thaliana egg cell-secreted-like protein (DUF1278) mRNA (see the first one in fig 6).
Figure 5.
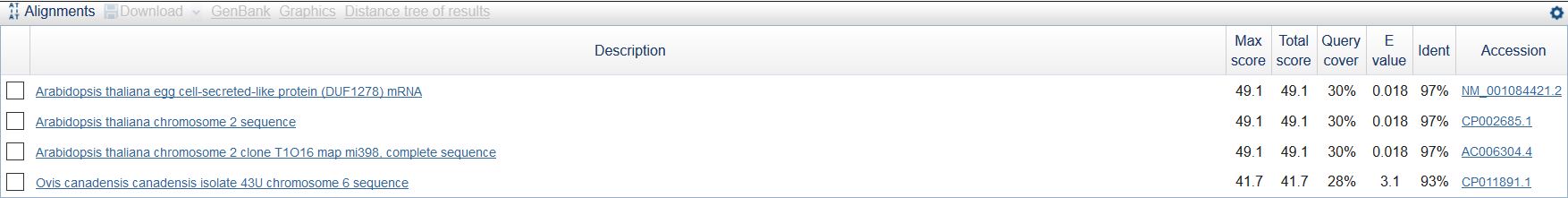
Figure 6.
| Term 3 |
| ← Pr 12-13 | Next term → |
© Darya Potanina, 2017