Сборка de novo
В четырнадцатом практикуме каждому студенту выдаётся код доступа, соответствующий запуску секвенирования в проекте по прочтению генома бактерии Buchnera aphidicola. Задача — собрать фрагмент генома и оценить результат.
Работа
Первым делом нужно скачать файл с прочтениями. Мне достался код SRR4240360. Так как все программы, используемые в дальнейшем, умеют работать с файлами .fastq.gz, архив не был распакован.
Подготовка прочтений к сборке включала две стадии триммирования (обрезка адаптеров и затем отбор по качеству и длине), исполненные шагами ILLUMINACLIP:my_adapters.fasta:2:7:7 (my_adapters.fa получен объединением файлов адаптеров с kodomo) и TRAILING:20 MINLEN:32. Был осуществлён мониторинг качества прочтений в изначальном файле и после каждой из операций (FastQC). Использованный сценарий можно скачать. Отчёты FastQC: 1, 2, 3. Диаграммы с качеством прочтения нуклеотидов сведены на рис. 1.
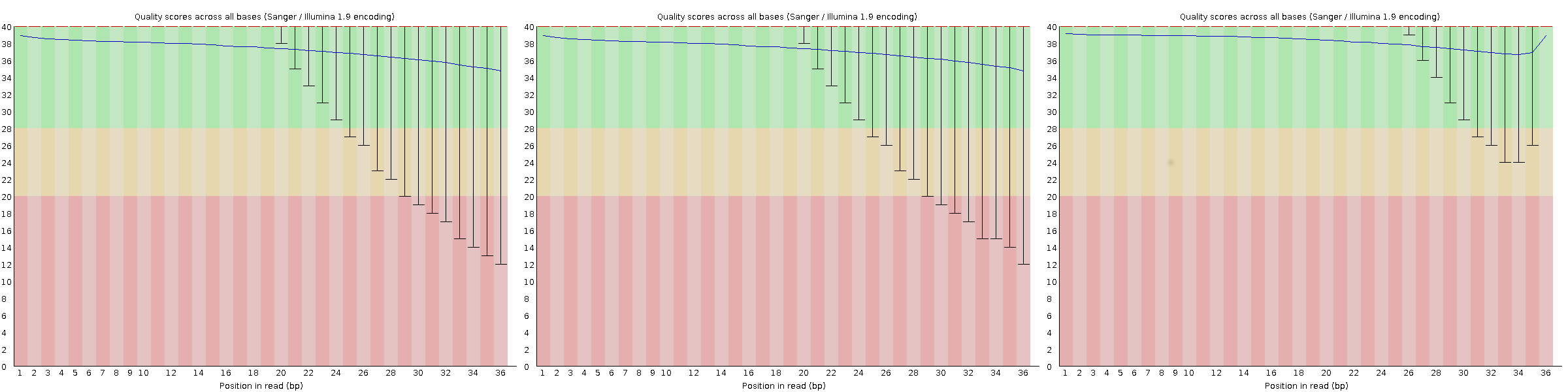
Кстати, на первом этапе подготовки чтений было удалено 41859 шт. (0,51 %), на втором — 297302 (3,62 %). До всех операций архив с чтениями занимал 193 МБ (если распаковать, 832 МБ), в итоге осталось 183 МБ (796 МБ). Интересно, что вплоть до конца в разделе Overrepresented sequences отчёта FastQC фигурировала в буквальном смысле последовательность поли-A, составляя до 23 % чтений.
Для сборки используем программу velvet (команды velveth и velvetg). Вызываем:
- velveth assem/ 31 -fastq.gz -short SRR4240360_trim.fastq.gz
- velvetg assem/
Здесь assem — папка для сборки, 31 — размер k-мера для построения графа, -fastq.gz — формат файла с чтениями, -short — указание на короткие непарные прочтения, SRR4240360_trim.fastq.gz — путь к самим чтениям.
Анализ сборки проводим на основе файла stats.txt, сгенерированного сборщиком. (Импортируем его в Excel). Оценим типичные значения покрытия (берём столбец short1_cov с полным покрытием, а в столбце short1_Ocov фигурирует покрытие только строго совпавшими прочтениями) как те, что находятся в пределах одного стандартного отклонения от среднего. Если брать величину для исходных данных, то она получается больше среднего (тогда предположим логнормальное распределение и будем искать логарифмы, отстоящие от нашего диапазона на величину, большую ln 5). С учётом того, что при сборке странно ожидать покрытие меньше 1, а нижняя граница диапазона оказалась меньше ln 5, получаем только аномально высокие покрытия. Правда, среди этих 6 контигов не оказалось достаточно длинных (хотя бы из 31 k-мера) для попадания в contigs.fa. Контиг номер 592 (из 595) имеет длину 0 (!) и формально бесконечное покрытие. Номер 557 — длину 1 и покрытие 134 953 (наверное, это тот самый поли-A). Следующие тоже недлинные, но покрытие на порядки меньше. Скорее всего, это какие-то регионы низкой сложности, ассоциированные с поли-A. Суммарная длина сборки (при учёте только contigs.fa) равна 682 349, N50 получаем равным 43 100, L50 — 5. Три самых длинных контига описаны в таблице 1.
| Номер в сборке | Длина | Покрытие |
|---|---|---|
| 1 | 113504 | 33,52546 |
| 5 | 83633 | 33,646065 |
| 4 | 64185 | 35,847323 |
Теперь сравниваем эти контиги с банковским геномом бактерии (проводим megablast, вводя AC CP009253). Результаты представим в таблице 2.
| Номер в сборке | Первый выровненный нуклеотид | Последний выровненный нуклеотид | Покрытие |
|---|---|---|---|
| 1 | 1 574 | 110 741 | 76 % |
| 5 | 3 755 | 79 107 | 74,95 % |
| 4 | 393 | 62 517 | 78,38 % |
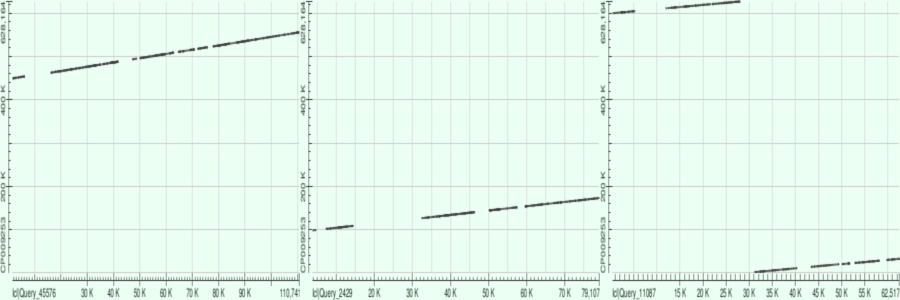
В целом видно, что контиги занимают значительную часть генома, а третий даже прошёл через нулевую координату. (Здесь не утверждается, что это необычно. Это важно и стоит упоминания, поскольку нулевая координата является выделенной на карте локального сходства, приведённой в качестве иллюстрации. В результате что угодно, минующее её, оказывается визуально разорванным, хотя при другом начале отсчёта не казалось бы таковым. Причём с точки зрения выравнивания нет разницы, откуда начинать нумерациию в кольцевой молекуле ДНК. Контиг не перестаёт быть контигом, хотя сходит с одной диагонали карты — если не считать диагонали замкнутыми через горизонтальные стороны карты. Вероятность контига пройти через нуль прямо пропорциональна длине его последовательно картированной части и равна отношению этой длины, выраженной в расстоянии между нуклеотидами в выровненном референсе, к полной длине референсной последовательности, поэтому, повторюсь, ничего необычного в прохождении через нуль нет.)
При выполнении задания 14 возникли неожиданные детали вроде поли-A прочтений, но, несмотря на это, результат получился осмысленный.