| Главная | Семестры | Проекты | Обo мне | Ссылки | Заметки | Назад к оглавлению |
Парные выравнивания белков. Применение алгоритмов парных выравниваний к белку PDAA_BACSU
Сравнение матриц аминокислотных замен
- Что означает числа (в частности число 62) после названия матрицы? В процессе создания матрицы необходимо как-то учитывать близкие, очень похожие или даже одинаковые последовательности. Для этого был придуман принцип кластеризации, это принцип таков, что эти близкие/одинаковые последовательности объединяют в один кластер для которого высчитывают собственные частоты встречаемости (средние по идентичности) и затем сравнивают другие последовательности с этим единым кластером, таким образом можно избежать погрешности при построении матрицы.
- Чем отличаются матрицы между собой, в том числе BLOSUM и PHAT? Для различных белков должны существовать различные матрицы ввиду того, что в зависимости от локализации в клетке белков, в них будут наиболее часты те или иные аминокислотные замены. Так, например, матрица BLOSUM показывает частоты аминокислотных замен для цитоплазматических белков (преимущественно гидрофильных), а для мембранных, в которых основной составляющей является липидный бислой, она будет уже не очень действенна (хотя для некоторых приближений ее можно использовать и используют), но все же лучше в таком случае использовать PHAT (эта матрица была изначально создана с использованием последовательностей мембраннных белков).
- Мною была созданна матрица аминокислотных замен на основе последних изменений базы данных BLOCKS (если интересно, вы можете прочитать о BLOCKS здесь, посмотреть эту матрицу с пошаговыми действиями и комментариями можно тут (Exel 97/00), тут (Exel 5.0) или тут (Open Office) ).
Вопросы и ответы по сравнению замены аминокислоты в различных матрицах аминокислотных замен:
Сравнительная таблица для глицина:
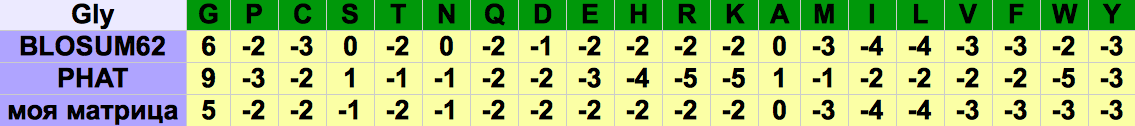
- Как отличаются в разных матрицах величины для замены глицина на самого себя? Почему? Глицин маленькая аминокислота и заменить его на самого себя в цитоплазматическом белке оказывается более выгодно, чем в мембранном.
- Как отличаются в разных матрицах величины замен выданной аминокислоты на аминокислоты близкие по химическим свойствам (если такие есть)? Почему? Близкими по химическим свойствам к глицину относятся гидрофобные алифатические аминокислоты (Ala, Met, Leu, Ile, Val, Pro). В BLOSUM62 и моей матрице значения совпали, т.к. я значально строила свою матрицу ожидая получение схожих с BLOSSUM результатов (из одинаковых соображений строила матрицу, но использовала данные из последней версии базы данных BLOCKS). В PHAT в целом более вероятны (кроме пролина) замены на другие алифатические аминокислоты, чем в BLOSUM и в моей матрице, очевидно, это связано с тем, что в PHAT рассматириваются мембранные белки и следовательно в них более вероятны замены одной гидрофобной аминокислоты на другую, чем в цитоплазматических белках, которые должны быть расстворимы в цитоплазме и которым позволяется иметь в основном только внутри глобулы неполярные участки, а следовательно гидрофобных аминокислот в целом меньше, чем гидрофильных. Вероятность смены глицина на пролин же меньше в PHAT, чем в моей матрице и BLOSUM, думаю, это объясняется тем, что пролин заставляет менять форму глобулы, стоит чаще всего в местах поворота полипептидной цепи, и замена маленькой аминокислоты глицина, которая почти никак не влияет на направление цепи на пролин означает изменение конформации белка, а форма мембранного белка (PHAT) чуть более важна чем цитоплазматического ввиду его “зажатости” в клеточной мембране, отсюда и более отрицательные значения, а “чуть более” потому, что изменение формы белка важно и для любого белка, в том числе и для цитоплазматической мембраны, у которого изменение формы может повлечь, например, изменение активного центра.
- Как отличаются в разных матрицах замены глицина на аминокислоты из других функциональных групп (хотя бы 2 примера)? Почему? Замена глицина на положительно заряженные аминокислоты (аргинин, лизин, гистедин) маловероятна (отрицательные значения), как в PHAT, таки в BLOSUM62 и моей матрице из-за того, что они довольно большие аминокислоты да еще и в целом положительно заряженные, так же для этих трех аминокислот общим является и то, что замена в BLOSUM62 и моей матрице более вероятна, чем в PHAT, я могу объяснить это тем, что в цитоплазме гидрофильные аминокислоты находятся снаружи, а в цитоплазматической мембране внутри (стараются не контактировать с липидным бислоем плазматической мембраны), а значит проще "пристроить" большие положительно заряженные аминокислоты в цитоплазме, следовательно такие и результаты. Отрицательно заряженные аминокислоты (аспарагиновую и глутаминовую кислоты) тоже не часто заменяют собой глицин, но чаще чем положительно заряженные. Почему? Относительно все равно же на положительно или отрицательно заряженную аминокислоту заменять маленький нейтральный глицин? Вот тут нам и приходят на помощь рассуждения о размере кислот. Положительно заряженные кислоты больше, чем отрицательные.
Сравнение выравниваний, полученных для коротких мутантов вручную и построенных классическими алгоритмами Нидлмана-Вунша и Смита-Ватермана
Программы needle и water - это программы из пакета EMBOSS. Программа needlе использует алгоритм Нидлмана-Вунша для глобального выравнивания, а water - Смита-Ватермана для локального. Главное отличие этих алгоритмов друг от друга в том, что алгоритм Нидлмана-Вунша "штрафует" за вставку gap вначале, а Смита-Ватермана - нет.
Параметры для проведения выравнивания:
- -gapopen - штраф за открытие gap (значение по умолчанию 10)
- -gapextend - штаф за удлинение gap (значение по умолчанию 0.5)
- -endopen - штраф за открытие gap в конце (значение по умолчанию 10)
- -endextend - штаф за удлинение gap в конце (значение по умолчанию 0.5)
- -datafile - используемая матрица (по умолчанию стоит BLOSUM62 для белковых последовательностей и EDNAFULL для нуклеотидных)
Сравниваются выравнивания 3 мутантов.
Результаты полученные вручную и общая характеристика мутантов:
| Изображение и номер выравнивания | 1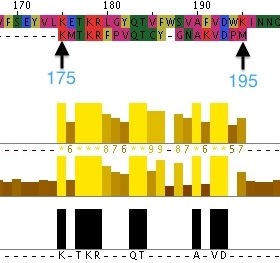 |
2.png) |
3.png) |
Параметры (подробнее тут):
|
|
|
|
| % идентичности | 30 | 39.1 | 60 |
| % сходства (по R) | 55 | 56.5 | 80 |
| вес по матрице BLOSUM62 | 19 | 31 | 57 |
Результаты полученные для needle:
мутант 1
PDAA_BACSU 151 YKITGKQDNLYLRPPRGVFSEYVLKETKRLGYQTVFWSVAFVDWKINNQK 200
|.|||...||.:.: |.||..
mutant1 1 ------------------------KMTKRFPVQTCYGN-AKVDPM----- 20
# Identity: 9/263 ( 3.4%)
# Similarity: 11/263 ( 4.2%)
# Gaps: 243/263 (92.4%)
# Score: 22.0
мутант 2
PDAA_BACSU 51 LIEKYDAFYLGNTKEKTIYLTFDNGYENGYTPK-VLDV-LKKHRVTGTFF 98
..|.| |||| :||:|....
mutant2 1 ----------------------------AKTVKVVLDVTMKKYRKWAV-- 20
# Identity: 9/265 ( 3.4%)
# Similarity: 11/265 ( 4.2%)
# Gaps: 247/265 (93.2%)
# Score: 19.0
мутант 3
PDAA_BACSU 1 MKWMCSICCAAVLLAGGAAQAEAVPNEPINWGFKRSVNHQPPDAGKQLNS 50
|||.:|.|||..|.||..||
mutant3 1 -------CCAVLLGAGGCNQPEAWSNE----------------------- 20
# Identity: 12/263 ( 4.6%)
# Similarity: 13/263 ( 4.9%)
# Gaps: 243/263 (92.4%)
# Score: 57.0
Результаты полученные для water:
мутант 1
PDAA_BACSU 175 KETKRLGYQTVF 186
|.|||...||.:
mutant1 1 KMTKRFPVQTCY 12
# Identity: 6/12 (50.0%)
# Similarity: 7/12 (58.3%)
# Gaps: 0/12 ( 0.0%)
# Score: 27.0
мутант 2
PDAA_BACSU 84 VLDV-LKKHR 92
|||| :||:|
mutant2 7 VLDVTMKKYR 16
# Identity: 7/10 (70.0%)
# Similarity: 9/10 (90.0%)
# Gaps: 1/10 (10.0%)
# Score: 27.0
мутант 3
PDAA_BACSU 8 CCAAVLLAGGAAQAEAVPNE 27
|||.:|.|||..|.||..||
mutant3 1 CCAVLLGAGGCNQPEAWSNE 20
# Identity: 12/20 (60.0%)
# Similarity: 13/20 (65.0%)
# Gaps: 0/20 ( 0.0%)
# Score: 57.0
Сводная таблица с результатами (для needle результаты пересчитаны для участка):
| Номер мутанта | Вручную | Needle | Water |
| 1 |
|
|
|
| 2 |
|
|
|
| 3 |
|
|
|
С помощью этой таблицы несложно заметить, что мои результаты по 1 и 2 выравниванию хуже, чем результаты полученные с помощью алгоритмов, а вот 3 выравнивание совпадает, а значит его можно считать удачным.
Сравнение выравниваний, полученных для полноразмерных мутантов вручную и построенных классическими алгоритмами Нидлмана-Вунша и Смита-Ватермана
Выравнивание последовательности моего белка относительно искусственно синтезированных (с помощью скрипта evolve_protein.pl, в котором можно изменять параметры) последовательностей (длиной 263 как и у моего белка). Значимо то, что эти параметры эколюционирования близки к реальным.
| Номер выравнивания | 1 | 2 | 3 |
Параметры:
|
|
|
|
| % идентичности | (263-21)/263*100%=~92.02% | (263-181)/263*100%=~31.18% | (263-22)/263*100%=~91.63% |
Как мы видим, при вероятности изменения остатка (параметр отвечающий за моделирование "ошибки" ДНК-полимеразы) равной 0.0001 и большом количестве поколений - 10000 (2 "мутант") процент идентичности резко падает в отличии от тех вариантов, когда либо меньше поколений (1), либо вероятность изменения остатка меньше (3). Эти результаты вполне логичны, ведь при большем количестве ошибок ДНК-полимереразы больше будет изменений и чем больше поколений пройдет чем больше у них будет "ошибок" предковых (которые были созданы при создании предыдущих поколений) и своих (которые произошли при создании их). Идентичность у 1 и 3 "мутанта" почти совпадает, ведь в одном случае у нас была большая "ошибка" полимеразы (в 10 раз), но зато мешьше поколений (тоже в 10 раз), а в другом "ошибка" была меньше, но зато поколений больше, причем эта разница между количеством поколений и процентом "ошибок" сопоставима - отсюда и такие результаты. Но как мы видим по этим результатам (1 и 3) вероятность "ошибки" ДНК-полимеразы вносит немного больший вклад в изменчивость, чем количество поколений.
Запуск программ needle и water для мутанта 2:
needle:
PDAA_BACSU 41 PPDAGKQLNSLIEKYDAFYLGNTKEKTIYLTFDNGYENGYTPKVLDVLK- 89
.|:|..||.|.|||.|||| .|.|||...:.:...|...|||..::.||
generations=1 40 APNAANQLYSGIEKSDAFY-PNIKEKIPGMWWAVEYSEAYTPNHIERLKW 88
PDAA_BACSU 90 KHRVTGTFFVT-------GHFVKDQPQLIKRMSDEGHIIGNH-SFHHPDL 131
...||....|| .|...| ||.||..|:.:. .:|||:
generations=1 89 DPYVTQIISVTTLSMPMCDHCYDD------RMFDELMIMTHRDGYHHPE- 131
PDAA_BACSU 132 TTKTAD-QIQDELDSVNEEVYKITGKQDNLYLRPPRGVFSEYVLKE---T 177
..:|| |:: .|.|...|..|||..|..|||...|: :|:|. |
generations=1 132 -AMSADHQLE---GSYNNYCYIRTGKPFNWCLRPFWRVW--FVVKAHEMT 175
PDAA_BACSU 178 KRLGYQTVFWSVAFVDWKINNQKGKKY--AYDHMIKQAHPGAI-----YL 220
|...|...|.|:|...|.|::..||.| |....||||| |: ..
generations=1 176 KISWYLCAFPSMATRAWAISHSPGKLYYHATFMQIKQAH--AVGCRKNLF 223
PDAA_BACSU 221 LHTVSRDNAEALDDAITDLKKQGYTFKSIDDLMFEK-EMRLPSL 263
.|..::...|||||..||.|:..||| |..:|:. .|..|.
generations=1 224 SHKRAQCEEEALDDIATDTKQGTYTF---DSGLFDGFAMNGPH- 263
# Identity: 95/294 (32.3%)
# Similarity: 124/294 (42.2%)
# Gaps: 62/294 (21.1%)
# Score: 192.5
water:
PDAA_BACSU 34 KRSVNHQPPDAGKQLNSLIEKYDAFYLGNTKEKTIYLTFDNGYENGYTPK 83
|||.| :.|:|..||.|.|||.|||| .|.|||...:.:...|...|||.
generations=1 34 KRSPN-KAPNAANQLYSGIEKSDAFY-PNIKEKIPGMWWAVEYSEAYTPN 81
PDAA_BACSU 84 VLDVLK-KHRVTGTFFVT-------GHFVKDQPQLIKRMSDEGHIIGNH- 124
.::.|| ...||....|| .|...| ||.||..|:.:.
generations=1 82 HIERLKWDPYVTQIISVTTLSMPMCDHCYDD------RMFDELMIMTHRD 125
PDAA_BACSU 125 SFHHPDLTTKTAD-QIQDELDSVNEEVYKITGKQDNLYLRPPRGVFSEYV 173
.:|||: ..:|| |:: .|.|...|..|||..|..|||...|: :|
generations=1 126 GYHHPE--AMSADHQLE---GSYNNYCYIRTGKPFNWCLRPFWRVW--FV 168
PDAA_BACSU 174 LKE---TKRLGYQTVFWSVAFVDWKINNQKGKKY--AYDHMIKQAHPGAI 218
:|. ||...|...|.|:|...|.|::..||.| |....||||| |:
generations=1 169 VKAHEMTKISWYLCAFPSMATRAWAISHSPGKLYYHATFMQIKQAH--AV 216
PDAA_BACSU 219 -----YLLHTVSRDNAEALDDAITDLKKQGYTFKS 248
...|..::...|||||..||.|:..|||.|
generations=1 217 GCRKNLFSHKRAQCEEEALDDIATDTKQGTYTFDS 251
# Identity: 87/235 (37.0%)
# Similarity: 110/235 (46.8%)
# Gaps: 37/235 (15.7%)
# Score: 210.5
На создание 1000 поколений E.Coli при делении клетки раз в 30 минут ей потребуется ~9.9658*30=298.97мин=4ч 59мин, так как увеличение количества поколений будет происходить как 2^(количество делений (в начальный момент она не поделилась, а значит 0 момент, когда одна E.Coli)) следовательно количество делений=log2(1000) и количество делений мы уножаем на 30 мин, так как 1 деление каждые 30 мин происходит. А на создание 10000 - 13.2877*30=398.63мин=6ч 39мин.