
|

|
ATP-syntase from Bacillus PS3
This structure is the first full Bacillus ATP synthase complex, that has received. Subunits of Bacillus PS3 ATP synthase, including subunit b bearing an N-terminal His tag, were expressed from a plasmid in E. coli. The complex was been extracted from membranes and subjected to cryo-EM analysis. It is a multi-subunit protein complex, which use the electrochemical gradient for producing ATP. In mitochondria the protein forms dimers and this interactions make mitochondria’s crystae
The enzyme was been researched in liposomes. It was been shown that ∆pH or∆Ꝕ alone may drive proton’s dislocation. F1 complex provides synthesis/hydrolysis ATP and F0 complex provides proton’s dislocation. Bacteria’s protein is simpler than mitochondria’s. The F1 region consists of subunits α3β3γδε, while the F0 region is usually formed by three subunits with the stoichiometry ab2c10.
ε-subunit inhibits enzyme in thermophilic bacillus when the ATP concentration is low and take the up-conformation. Subunit take the down-conformation when the ATP conformation is high.
Ionic interactions, short secondary structure’s loops and closely spaced atoms in hydrophobic pockets provide protein’s stability and active in thermophilic bacteria. Ionic interactions just have shown for ATP synthase. Conceivably, its interactions with subunit’s interactions together stabilize complex. [1]
On F1 complex there are 3 catalytic and 3 non-catalytic sites. On α-subunit there is the non-catalytic sites, on α-subunit and β-subunit both there is the catalytic site. A large part of its is located on β-subunit. There is a Mg/ATP-complex in non-catalytic site. The protein is in inhibit conformation. [2]
There is a regulatory α-helix in ε-subunit. In E. Coli there are two parts of this α-helix, which are broken by a loop with 10 aminoacids. This loop contacts with γ-subunits and presumably can stabilized the up-conformation. This can be a reason for E. Coli inhibition, which doesn’t depends on ATP concentration.
Conceivably, ε-subunit in up-conformation does not inhibit the ATP synthesis but inhibits the ATP hydrolysis. The ε- and γ-subunit’s collision may block the rotation in hydrolysis’s process.
A channels for proton’s transport are same with mitochondria’s and chloroplast’s. A cytoplasmatic half-channel forms by a-subunit and c-ring, a periplasmatic half-channel forms by α-helixes number 1, 3, 4, 5 on a-subunit and by space between α-helixes number 5 and 6 on c-ring. The channel’s diameter is 1.4Å. A proton joins with Glu56 on c-ring, than rotates in hydrophobic membrane and than joins with Arg169 on a-subunit. [1]
This enzyme has a lot of inhibitors; the main are ADP, ε-subunit and phosphate. The main type of inhibition varies in organisms. The protein from E. Coli can inhibit by phosphate and the protein from Basillus PS3 can inhibit by ADP. It probably can relate on bacteria’s environment: Basillus PS3 are free-living aerobes, E. Coli are optional anaerobes. [2]
Contact’s atlas
Ligand’s interactions
6N2Y
|
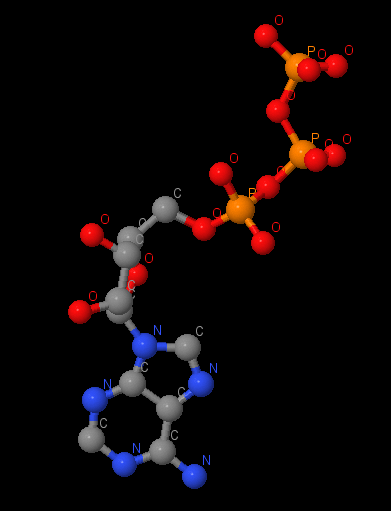
Name: ATP Gross formula: C₁₀H₁₆N₅O₁₃P₃ UIPAC name: [[(2R,3S,4R,5R)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate Molecular weight: 507,18 g/mole PubChem ID: 5957 |
|
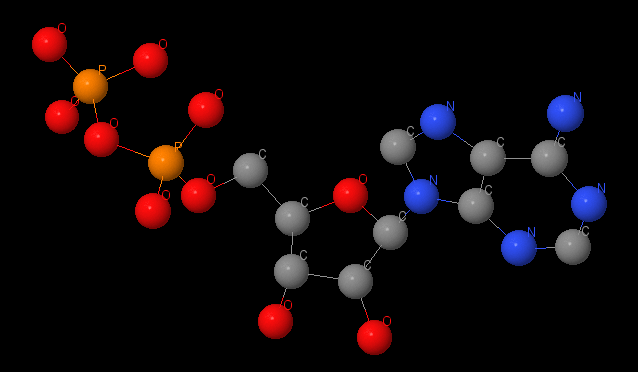
Name: ADP Gross formula: C₁₀H₁₅N₅O₁₀P₂ UIPAC name: 9-{5-O-[Hydroxy(phosphonooxy)phosphoryl]-β-D-ribofuranosyl}-9H-purin-6-amin Adenosin5'-(trihydrogendiphosphat) Molecular weight: 427,203 g/mole PubChem ID: 6022 |
|

|
Name: magnum Gross formula: Mg UIPAC name: Magnesium Molecular weight: 24,304 g/mole PubChem ID: 5462224 |
6N2Z
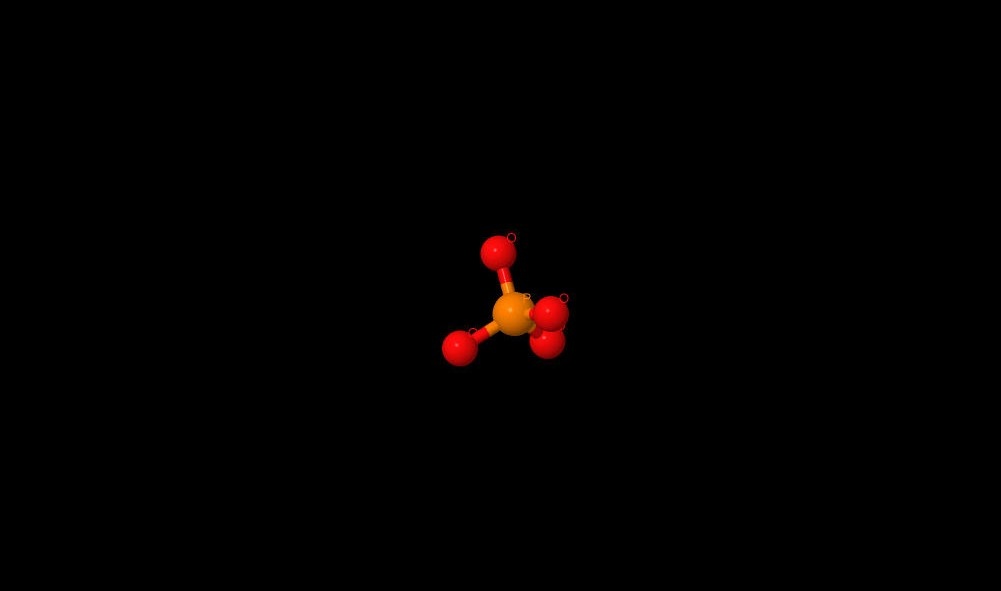
|
Name: phosphate Gross formula: PO4 3- UIPAC name: phosphate Molecular weight: 94.9714 g/mole PubChem ID: 1061 |
Ligand’s interactions with protein
6N2Y
.png)
|
There is ATP in 3 non-catalytic sites. We can see 3 conformations of catalytic sites: open, close and half-closed (with ADP). |
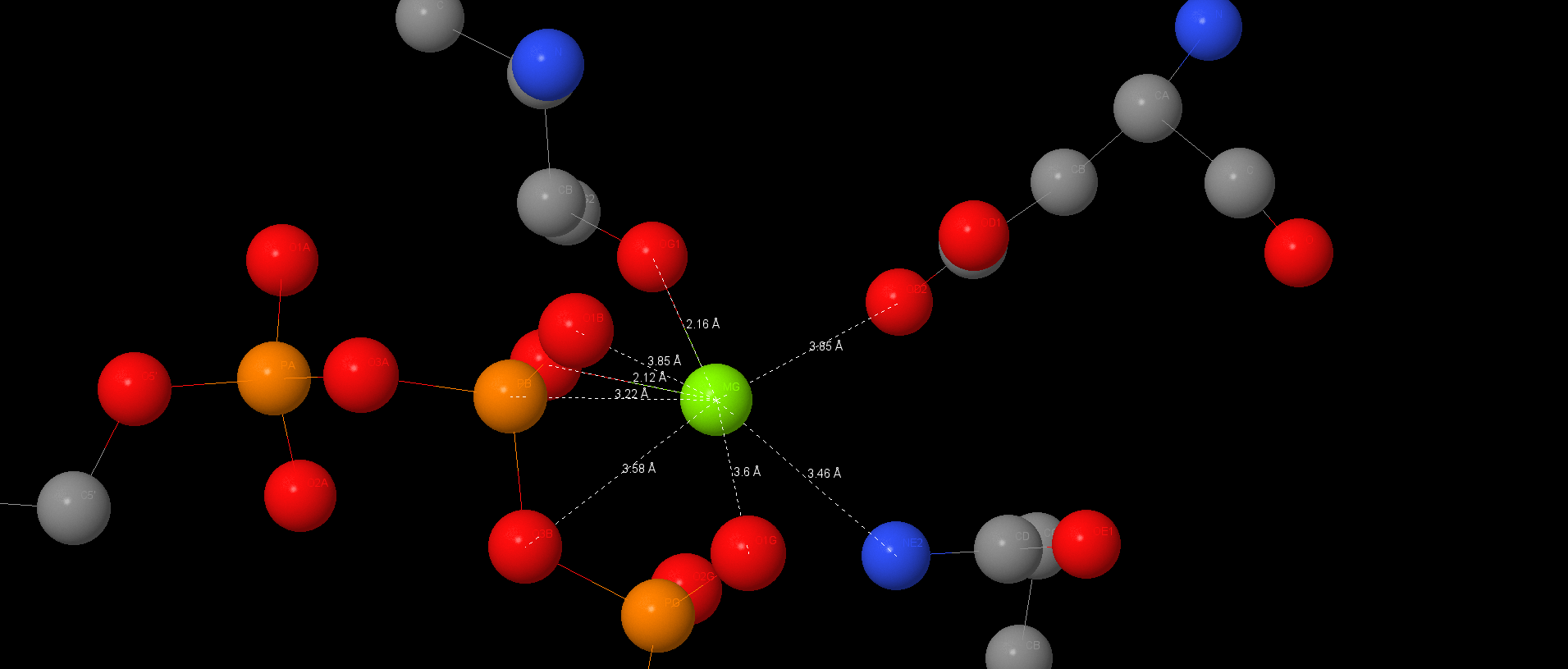
A magnum on α-subunit has a coordination interaction with ATP and may has a coordination interactions or salt-bridges with 176Thr, 261Asp, ATP by oxygen and with Glu200 by nitrogen. We can also watch ATP on other α-subunit. Magnum here has the coordination interactions with Thr176 and ATP. Conceivably, there are hydrogen bonds between oxygen in ATP and nitrogen in Ser177 and Glu172. Salt-bridges may be between Gly174, Lys175, Glu172.
It 7Å distance there is stacking-interaction between adenine in ATP and Phe349. Pi-systems are connected by slats. This interaction isn’t such profitable then connecting over each other. It may stabilizes the ligand in non-catalytic site.
| The radical of Arg354 is located in ~4.1Å from phenylalanine ring and ~5.1Å from adenine. It interacts with phenylalanine ring (pi-interaction) and it may stabilizes ATP in non-catalytic site. |
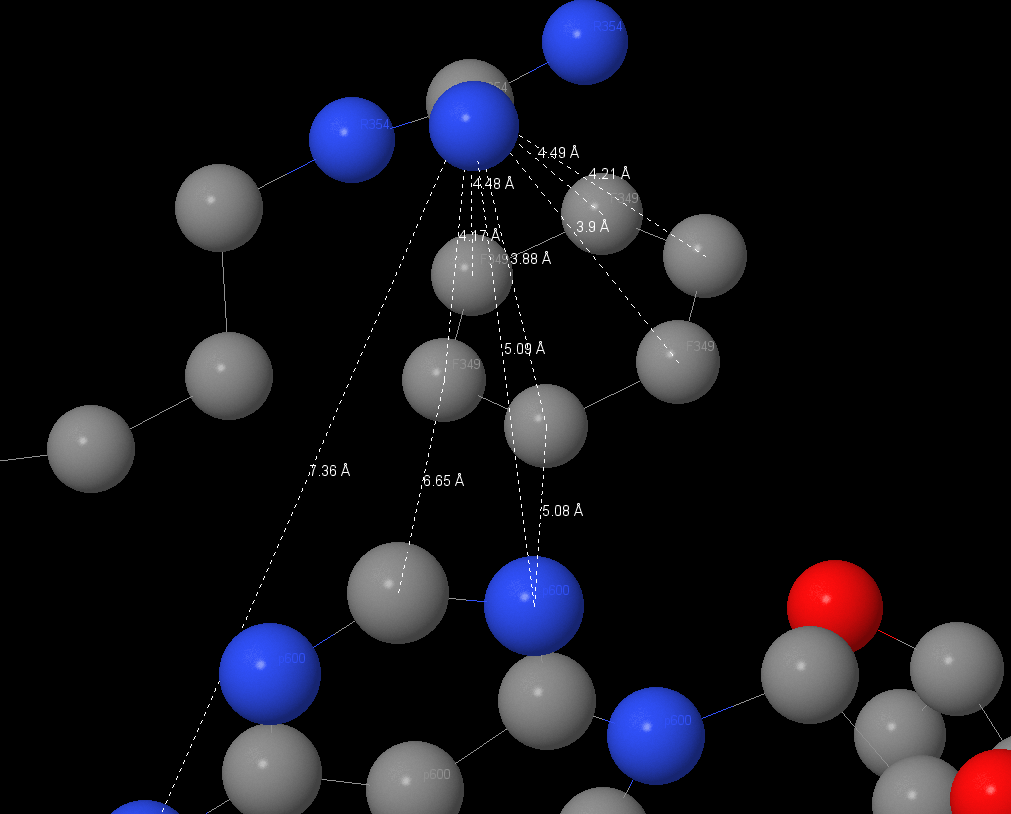
|
In half-close site ADP, the product of hydrolyze, is also stabilizes by stacking-interactions with Tyr341 and Phe420. ADP is also connected with Magnum by coordination interactions and Mg is connected with protein too.
Ligands guested on base of protein’s functions and then found by Jmol. After in Jmol I found ligand’s environment atoms, made measurements and suggested connects.
6N2Z
| PO4 3-, a product of ATP hydrolyze, has found in the half-closed site. It can inhibit enzyme in E. Coli and it may connects with aminoacids by hbonds. The connection lengths are: 2.51Å (with Gly161), 3.08Å (Ala160). |
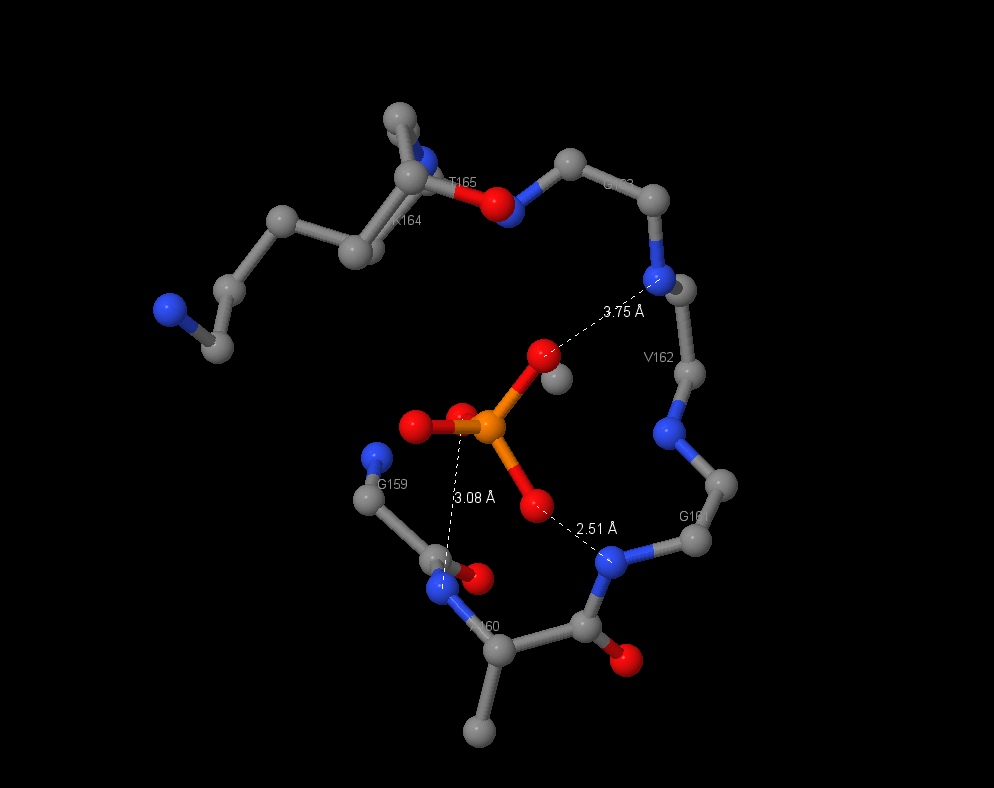
|
Interactions in protein
6N2Y
Stacking-interactions
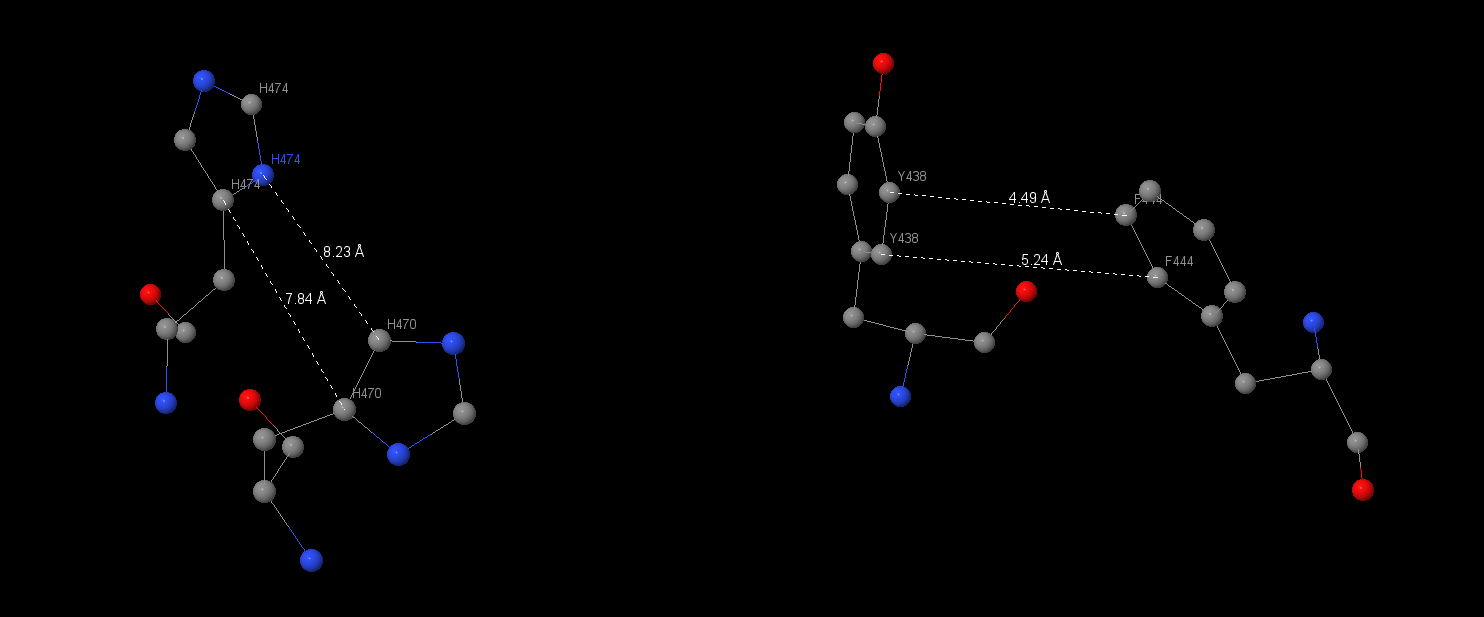
This type of interactions is very common in ATP synthase. It connects 2 or 3 aromatic rings. The stacking interactions were been found on α-subunit between Hys470 and Hys477 and between Tyr438 and Phe444.
| Connecting’s lengths between aromatic rings were been measured. In the first interaction they drawn up 7.84Å and 8.32Å, in the second – 4.49Å and 5.24Å. Another stacking-interaction has found in α-subunit Phe51, Trp54, Phe58, Phe219. I also measured connects between 51 and 54 aminoacids: 6.88, 5.77, 4.92Å, between 54 and 58: 5.37, 5.68Å, between 58 and 219: 3.47, 3.87Å. |

|
| I have also measured angles between aromatic rings. They are close to 90⁰. Connects between 51 and 54 aminoacids are 134.7⁰ and 114.3⁰ respectively; between 54 and 58 – 83.1⁰ and 86.8⁰; between 58 Åand 219: 72.4⁰ and 85.4⁰. | |
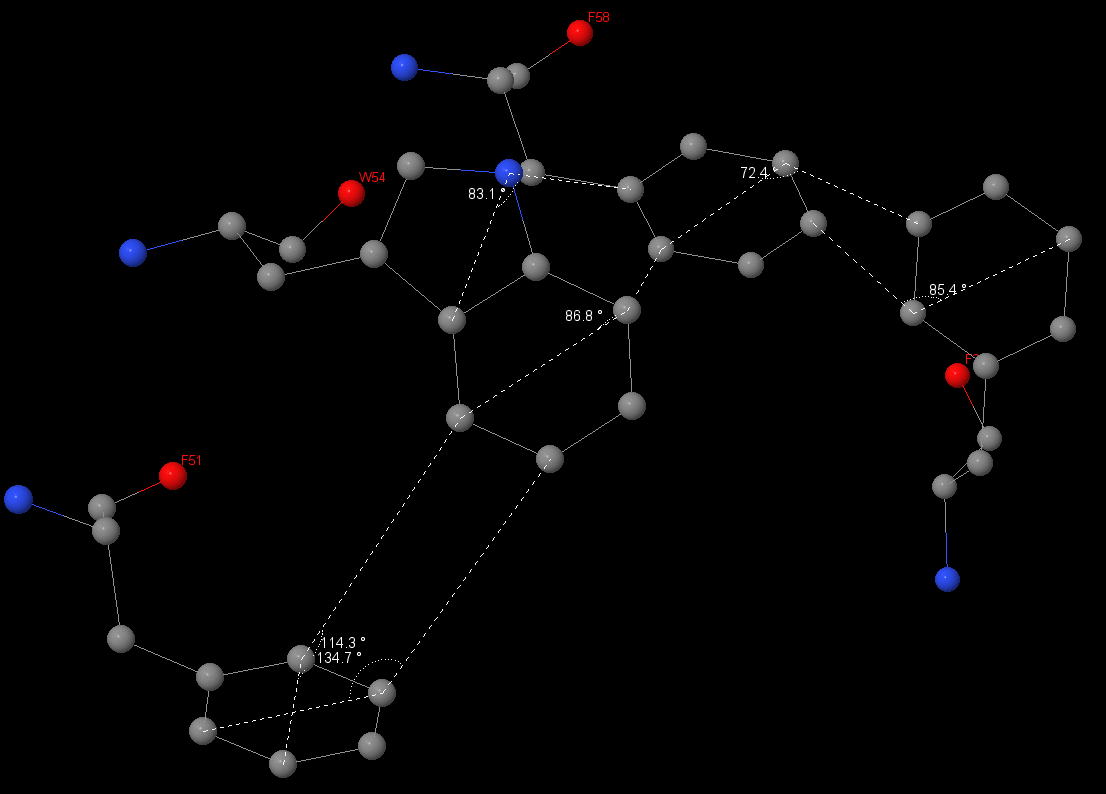
|
|
Stacking-interactions has been found by selecting all aminoacids with aromatic rings. After I chose aminoacids, which were placed near and over each other. Also, I found a longer system (4 aminoacids).
Hydrophobic pocket
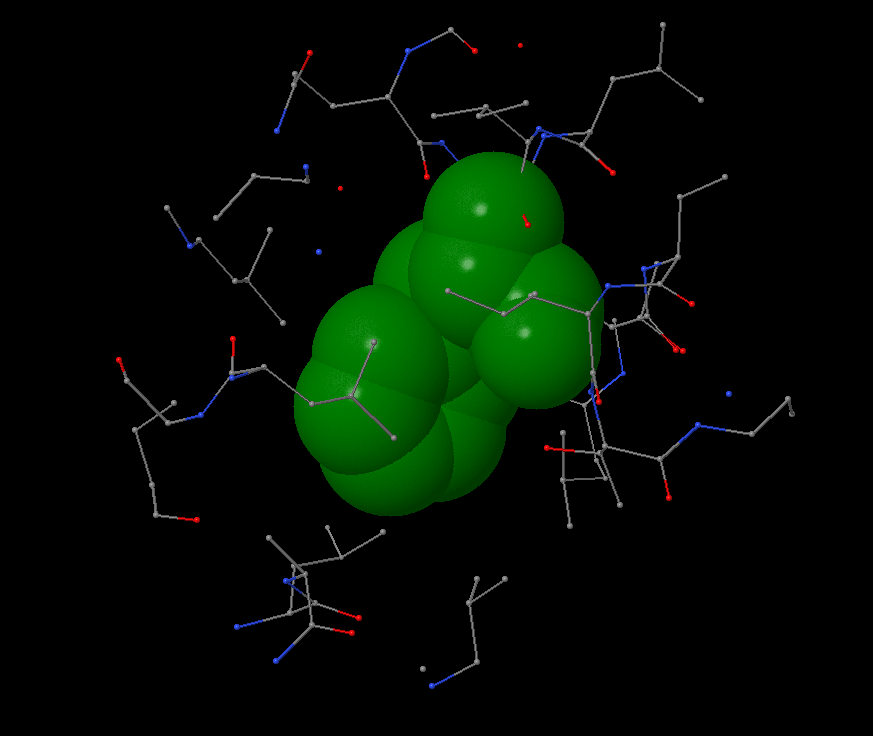
Hydrophobic pocket has been found near the Phe65 on c-subunit. It is surrounded by leucine and isoleucine from it’s and nearby subunits. In 4Å distance the first atoms of aromatic residue occur. Phenylalanine is surrounded by other atoms in 6Å distance. No atom can take place in this pocket: the average atom radius is 1.7Å, the distance between atoms in pocket is ~0.5-1.5Å.
There are a lot of hydrophobic aminoacids in c-ring, as well as hydrophobic pockets because c-ring is the membrane part of the protein. An average distance between non-covalent introductions in protein is 4.5-5Å.
|
I found a big hydrophobic aminoacid, chose the most beautiful and started to work with “within” command. |
Structure hbonds
Hbonds between elements of the secondary structure have been found on ε-subunit. A connect between Val6 and Asp18 (length 3.69Å, can be a salt-bridge) unites β-sheet and loops.
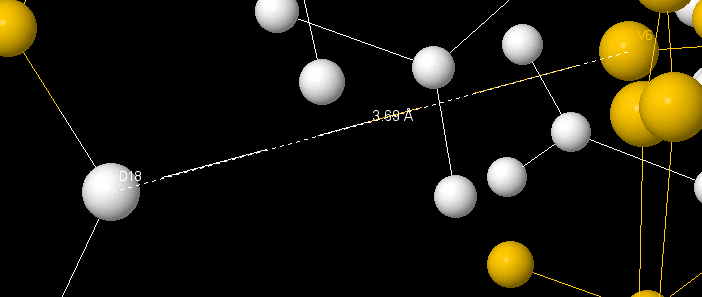
|
|
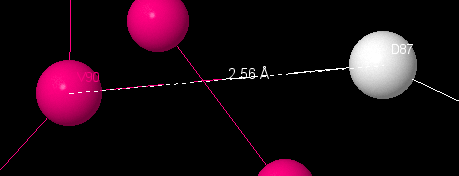
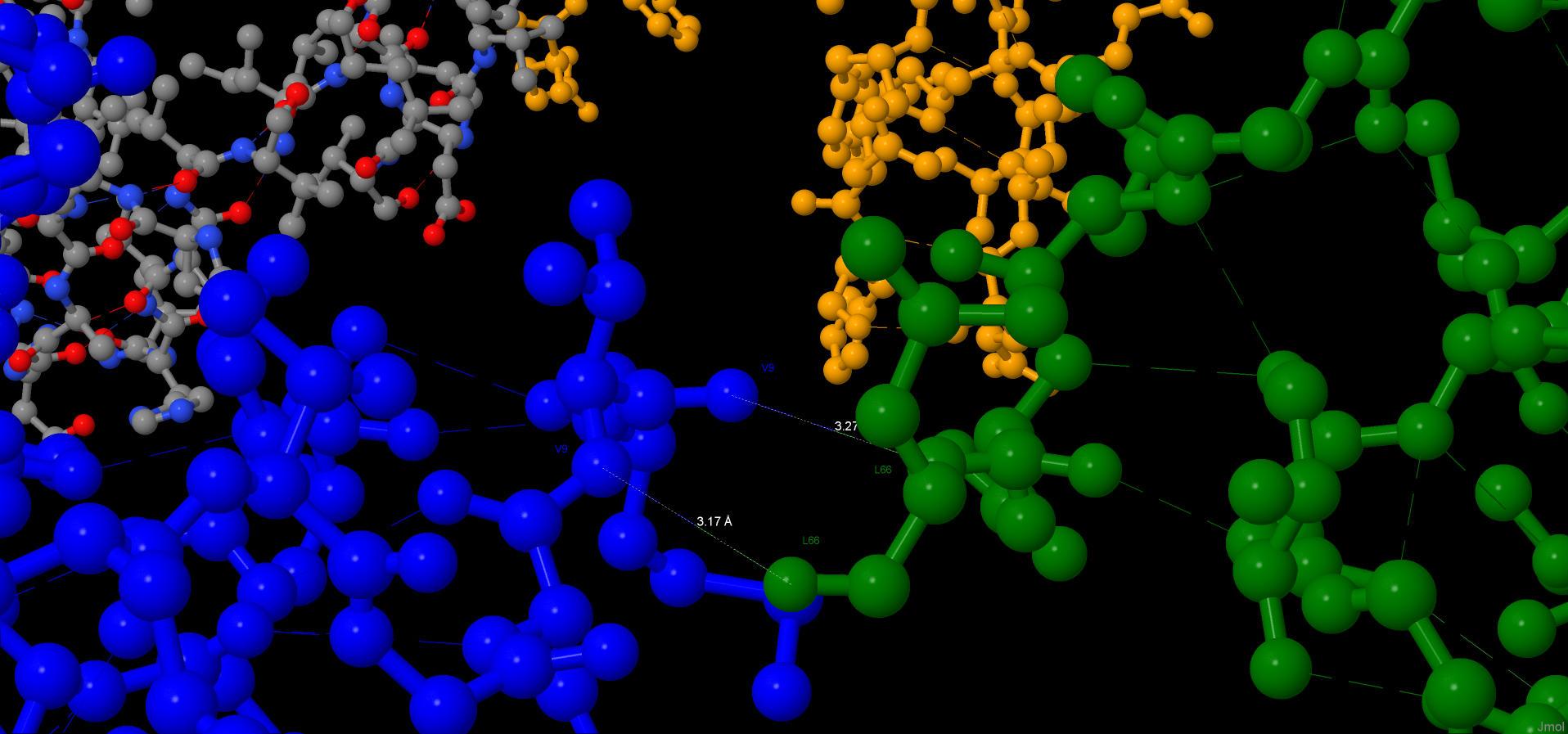
There is a hbond between Val90 and Asp87, which connects α-helix and loop. It’s length is 2.56Å. There are hbonds between Ala79 and Ile77 (3.17Å) and Ala79 and Val9(3.48Å).
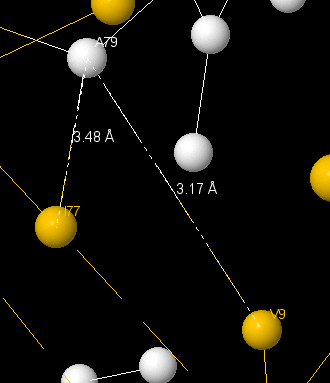
All connects have been found in ε-subunit, which regulates protein’s activity. On this pictures β-sheets are yellow, α-helixes are pink and loops are white.
This interactions have been found by watching colored protein’s subunits with calculated hbonds. Hbonds have been found manually: they are two-colored.
Hbonds in secondary structure
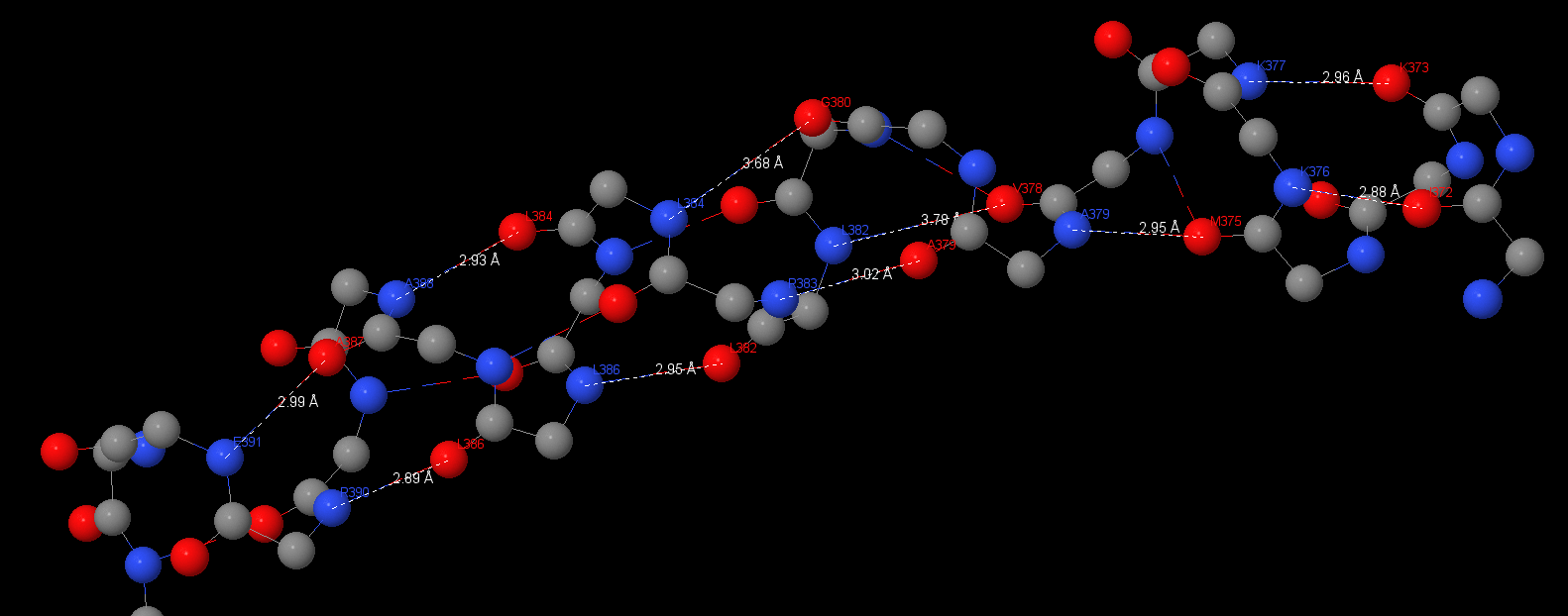
This protein has a lot of 3.10 α-helixes and β-sheets. Angles and distances are typical for hbonds:
391Glu(N) – Ala387: 2.99 Å;
Asp390(N) – Lys386: 2.89 Å, 150.9°;
Ala388(N) – Lys384: 2.93 Å, 153°;
Lys386(N) – Lys382: 2.95 Å, 150.1°;
Asp383(N) – Ala379: 3.02 Å, 154.8°;
Ala379(N) – Met375: 2.95 Å, 160.5°;
Asp376(N) – Ile372: 2.88 Å;
Asp377(N) – Asp373: 2.96 Å, 148.6°;
The average angle is 153⁰, the average distance is 2.95Å. Hbonds distances aspires to ideal 3Å and angle deviates from ideal for ~30⁰. This research was the easiest because there are a lot of these elements. We can also see some long α-helixes in b-subunits and ε-subunit.
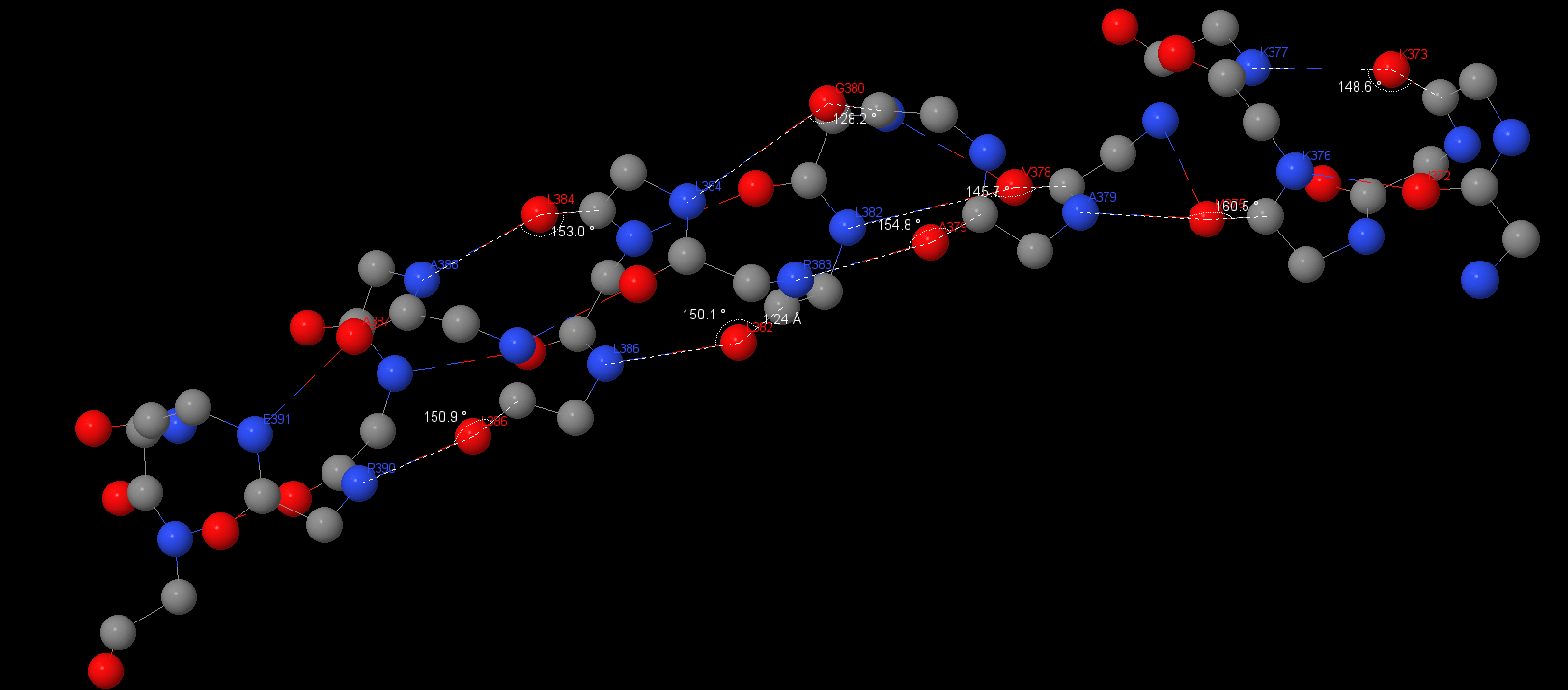
S-S
This type of interaction isn’t exist because there is only one cysteine.
Salt-bridges
Salt-bridges have been found in β-subunit in F1 complex. I have investigated interactions between 3 aminoacids: 2 glutamine acids and lysine.
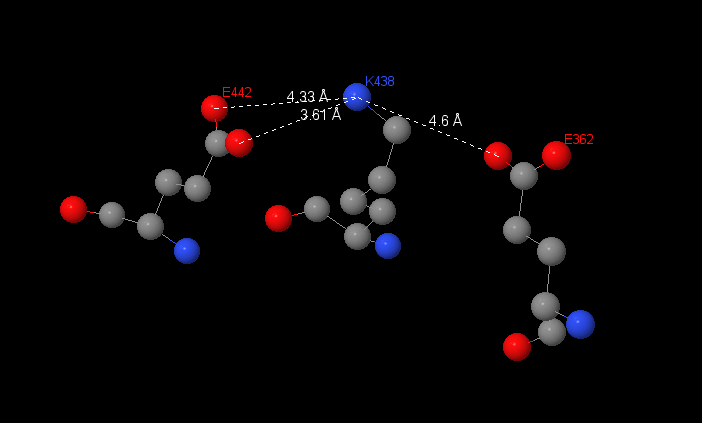
|
Salt bridges don’t have clear boundaries, which hbonds have. Because of this I can suggested interactions between radicals in Glu442, Glu362 and Lys438. |
The distance between this aminoacids are 3.61, 4.33 and 4.6Å. But the interactions are as bigger as the distance is shorter, so I assume the salt-bridge between Glu442 and Lys436 because the distance between them is the shortest.
I found salt-bridges by selecting aminoacids, that have charged when the pH is normal. After I chose aminoacids, which are close and measured distances.
6N2Z
Structure hbonds
β-sheets can be connected with hbonds, but it isn’t common for ATP synthase. The average distance between N-O is 3.13Å, the average angle N-O-C is 123⁰. The distance is close to ideal, but the angle is less than theoretical.
Hydrophobic pocket
|
In the 4-5Å distance from phenylalanine the other atoms start to “close” it. The characteristic distance between non-covalent introductions in protein is 4.52Å (measurements 4.36, 4.68, 5.5Å). |
Jmol-applet
|
|
Download scripts
Literature
1. Hui Guo, Toshiharu Suzuki, John L Rubinstein. «Structure of a bacterial ATP synthase», eLIFE 2019
2. A. S. Lapashina and B. A. Feniouk, ADP-Inhibition of H+-F0F1-ATP synthase, Biochemistry 2018
Author contributions
Zubareva Valeria looked for interactions in 6N2Y structure, described them, wrote Jmol-scripts (all work with 6N2Y structure). Also she described protein, wrote html-page and translated page to English.
Shilaeva Maria found interactions in 6N2Z structure, described them and wrote scripts (all work with 6N2Z structure).
Reasons for choosing 2 structures
In eLIFE (2019) there were 3 ATP synthase structures, which are different in catalytic state’s conformation. Because our group was compiled by people, which has absolutely different working capacity. Zubareva Valeria described all types of interactions by the 2 of March. So she suggested a new working-plan and gave another structure, which contains a new ligand – phosphate ion.
Reasons for another page, the third author
Zubareva Valeria was coorditating the whole work, so she asked everyone to do all by sunday evening, because she wanted to have enough time for correcting all material. But the third author didn't give me material, so we have finished work without her. But after deadline she done the work, so I think I have to put here her work on the link. Thanks for understanding.