Задание 1
Анализ качества чтений
Для выполнения использовалась хромосома 7 - fasta-файл и файл с одноконцевыми чтениями chr7.fastq).
Применим команду FastQC.
Рисунок 1 FastQC
В результате получены архив chr14_fastqc.zip и файл в форматеhtml, визуализирующий информацию о чтениях.
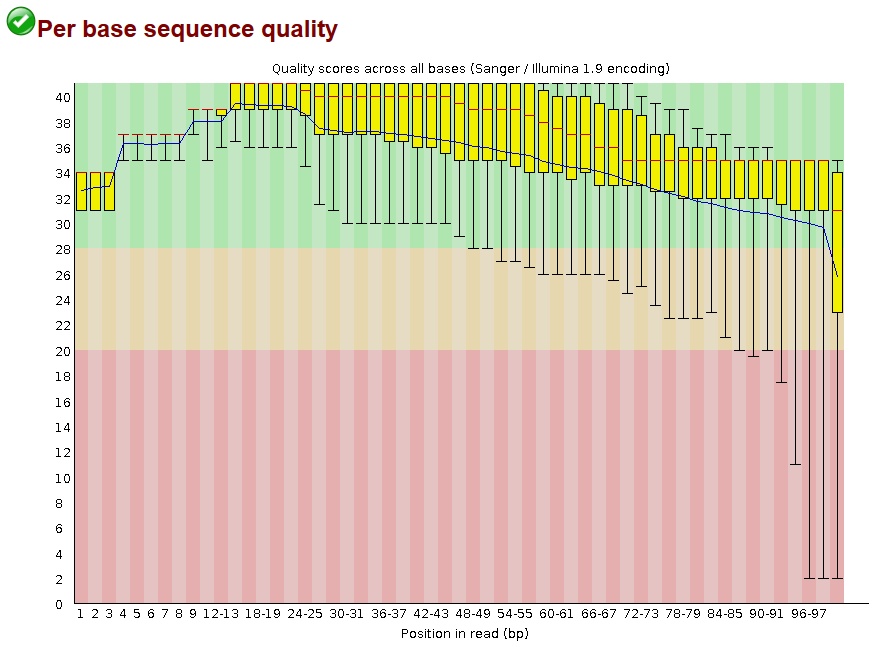
Рисунок 2 Картинка "Per base quality"
Видно, что конец ридов прочитан гораздо хуже их основной части
Это объясняется тем, что к некоторой итерации при секвенировании происходит накопление ошибок
На рисунке 3 представлено поле "Basic Statistics"в котором содержится информация о количестве, длине, GC-составе чтений и виде секвенирования.
Можно узнать конкретную версию секвенатора Illumina (Illumina 1.9),общее число чтений (3752),диапазон длины ридов (30-100)
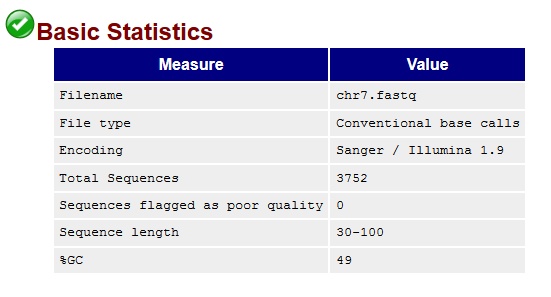
Рисунок 3 Таблица "Basic Statistics" (до чистки).
Очистка чтений
Очистим чтения при помощи программы Trimmomatic. Отрежем с конца каждого чтения нуклеотиды качеством ниже 20, оставив только чтения длиной не ниже 50 нуклеотидов

Рисунок 4 Trimmomatic
Таким образом, после чистки осталось 3650 (97,28%) ридов, то есть было удалено 102 (2,72%) чтений.
Запустим вновь FastQC
Рисунок 5 Команда, запускающая программу FastQC (после чистки).
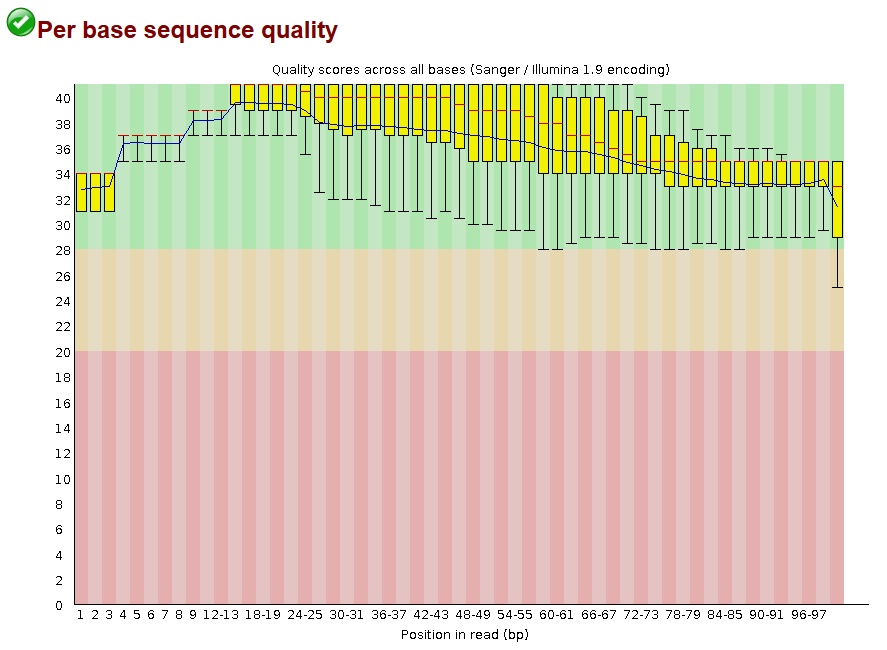
Рисунок 6 Качество чтений после чистки.
Хорошо видно, откаких ридов избавился Trimmomatic: исчезли чтения, у которых на концах были нуклеотиды плохого качества.
Отсутствуют позиции, значения качества которых находятся в красной области, и только последняя позиция расположена в желтой, следовательно цель отбора более качественных чтений достигнута.
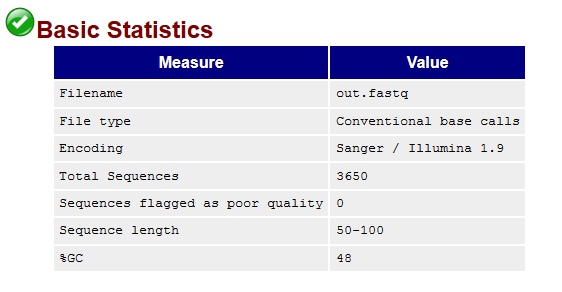
Рисунок 7 Таблица "Basic Statistics" (фрагмент выдачи программы FastQC после чистки). Заметим, что были удалены чтения длиной меньше 50 нуклеотидов, и теперь минимальная длины рида равна 50.
Картирование чтений
С помощью программы BWA надо было откартировать очищенные чтения.
Проиндексируем референсную последовательность хромосомы 7
Рисунок 8 Результат работы команды bwa index, индексирующей референсную последовательность
Построим выравнивание прочтений и референса в формате .sam
 Рисунок 9 Команда bwa mem, строящая выравнивание прочтений и референсной последовательности
Рисунок 9 Команда bwa mem, строящая выравнивание прочтений и референсной последовательности
Анализ выравнивания
Переведём выравнивание чтений с референсом в бинарный формат .bam,
Запустим команду samtools view с опциями -b и -о

Рисунок 10 Результат работы команды samtools view
Отсортируем выравнивание чтений с референсом (получившийся после картирования .bam файл) по координате в референсе начала чтения с помощью команды samtools sort с опцией -T с последующим указанием файла, куда складываются временные файлы.

Рисунок 11 Результат работы команды samtools sort
Далее проиндексируем отсортированный .bam файл

Рисунок 12 Результат работы команды samtools index
Получим информацию о том, сколько чтений откартировалось на геном

Рисунок 13 Результат работы команды samtools idxstats
Таким образом, в моем случае картировалось 3648 ридов
Анализ SNP
Произведём поиск SNP для их последующего анализа.
Для этого был создан файл со списком отличий между референсом и чтениями в формате .vcf.
Рисунок 14
Рисунок 15
Опишем три полиморфизма из .vcf файла.
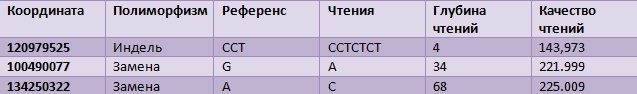
Рисунок 16 Результат работы команды samtools idxstats
С помощью программы annovar были проаннотированы только полученные snp. Были использованы базы данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
Были удалены все индели, после чего файл diff.vcf был переведен в формат .avinput для успешной работы с annovar.
Отчет о полученных snp
Было получено 3 инделя и всего 31 snp
15 (почти половина от общего числа) snp имеют покрытие больше 10, что считается хорошим покрытием и имеют высокое качество чтений.
Категории: exonic, splicing, ncRNA, UTR5, UTR3, intronic, upstream, downstream, intergenic
exonic -4
intronic - 24
UTR3 - 3
SNP попали в 3 гена:
ACHE - кодирует белок, необходимый для дезактивации ацетилхолина в синаптической щели и перехода клетки-мишени в состояние покоя (например, для расслабления мышечной клетки).
WNT16 - экспрессия данного гена отвечает за устойчивость раковых клеток к химиотерапии
AKR1B15 - Белок, который кодирует данный ген, принадлежит к aldo-keto reductase family
Данные приведены в файле exonic_variant_function, полученном при аннотации по refgene и продублированы в файле
26 snp имеют rs
Среди аннотированных snp находится
0 с частотами от 0.8 до 1 (очень распространенные).
18 с частотами от 0.3 до 0.7 (достаточно распространенные).
0 c частотами от 0.1 до 0.3 (не очень распространенные)
7 c частотами менее 0.1 (редкие)
SNP в гене ACHE вызываeт риск сахарного диабета, а SNP в гене WNT16 отвечает за плотность минералов кости и толщину коры головного мозга
Файл, содержащий сводную таблицу аннотаций и использованные команды.
Ссылки
© Козлова Анастасия, 2015