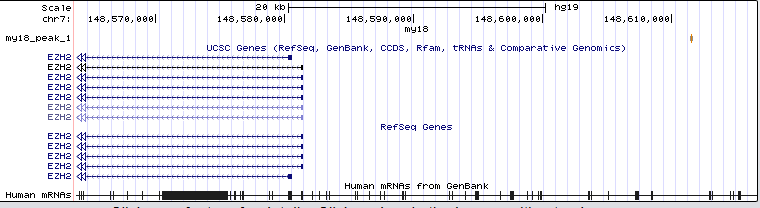
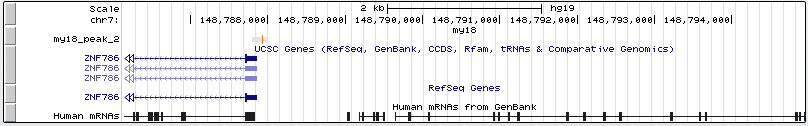
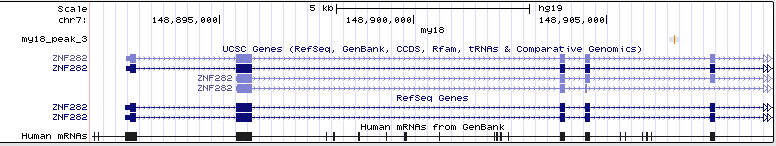
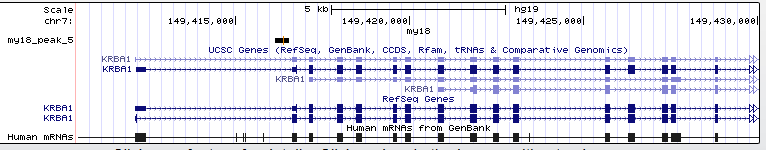
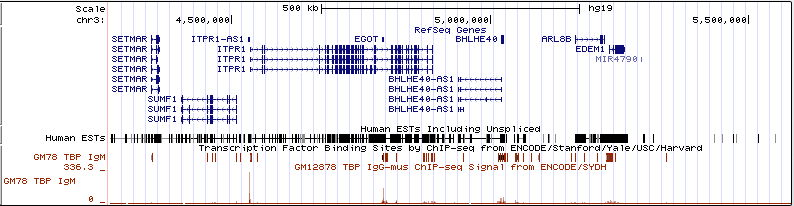
Блок 3: сигналыЗадание 1: Сайты рестрикции-модификацииДля анализа сайтов рестрикции-модификации (РМ-сайтов) были выданы идентификатор CP002367.1 и имя вида Selenomonas sputigena По идентификатору был распознан геном бактерии Lactococcus lactis (Bacteria; Firmicutes; Bacilli; Lactobacillales; Streptococcaceae; Lactococcus; Lactococcus lactis). Бактерия широко используется в пищевой промышленности в производстве сыров благодаря способности сбраживать сахара в молочную кислоту. Selenomonas sputigena (Bacteria; Firmicutes; Negativicutes; Selenomonadales; Selenomonadaceae; Selenomonas; Selenomonas sputigena) населяет ротовую полость человека. Поскольку в ротовой полости содержится лизоцим - фермент, расщепляющий клеточную стенку бактерии - sputigena должна быть к нему устойчива. Если лизоцимы фагов похожи на человеческий (к сожалению, нет времени посмотреть), то бактерия будет иметь и защиту от фагов. А значит, удалять большое колическтво РМ-сайтов ей нет смысла.Архивированные геном lactis и контиги sputigena вместе со списком РМ-сайтов были отданы на обработку сервису calculate compositional bias. Результаты (CP002637.1.fasta.gz и Selenomonas_result.tsv соответственно) были загружены в RM_sites.xlsx. В Excel посчитано число подпороговых (меньших 0,78) отношений ожидаемого числа РМ-сайтов к реальному и сделан список подпороговых РМ-сайтов. Затем оба списка подпороговых сайтов, sputigenia и lactis, были сопоставлены. У sputigena не оказалось ни одного удалённого РМ-сайта, который не был бы удалён у lactis(общие отсутствующие РМ-сайты обведены зелёным и выделены звёздочкой): 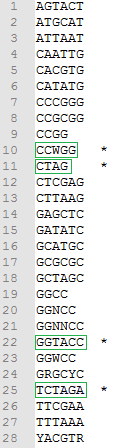 Задание 2: последовательность Шайн-ДальгарноПоскольку скрипт features2CDSs.py по отгонке CDS в xls с моей бактерией работать отказался (формат features у неё довольно странный), взяла любимый Deinococcus radiodurans, 1ую хромосому. Файлы:Deinococcus.fasta - 1ая хромосома, Deinococcus.ft - features. После отработки скрипта ( python features2CDSs.py -i Deinococcus.ft -o Deinococcus.xls) получен Deinococcus.xls, в котором с помощью "удалить дубликаты" были сначала удалены все "hypothetical protein" и "conserved hypothetical protein" (а только они в столбце product и повторяются). Потом с помощью "если" был создан столбец с нулями в тех строках, где max_coord-min_coord<900, то есть длина гена менее 900. И уже оттуда удалены совпадения по нулям. Итого остались прелестные 606 генов, которые будем смотреть.Для отбора взяли область в 17 кодонов от стартового: если ориентация прямая, то берём область [min_coord-16, min_coord], если обратная - то берём [max_coord, max_coord+16]. Результат отбора с заголовком из Excel копируем в txt (заголовок комментим диезом!) и отдаём скрипту fragments2fasta.py ( python fragments2fasta.py -i select17.txt -f Deinococcus.fasta -o select17.fasta). На выходе получаем fasta-файл с 17-нуклеотидными участками, где будет искаться сигнал Шайн-Дальгарно: AGGAGG на прямой цепи и CCUCCU на обратной.Для выполнения поиска с помощью MEME надо: - задать фаста-файл с координатами 17-нуклеотидных отрезков (Input the primary sequences), - выбрать, сколько раз может мотив встречаться на каждой из выравниваемых последовательностей (Select the site distribution), - выбрать число мотивов, которые будут искаться в выравнивании (Select the number of motifs)- об этом немного подробнее будет ниже, - отредактировать Advanced options, задав минимум и максимум столбцов в паттерне (длину мотива то есть) и поиск мотива только на заданной цепи или на обеих (Can motif sites be on both strands? (DNA/RNA only)). Итог - файл с результатами MEME. При поиске только одного мотива был обнаружен не Шайн-Дальгарно - возможно, его стоит исследовать в дальнейшем, если будет время, поскольку для Deinococcus radiodurans он более специфичен. При поиске 3х мотивов Шайн-Дальгарно был найден вторым, а третьим стал мотив, фактически идентичный первому, но смещённый на одну позицию влево в выравнивании. Параметры поиска мотива:  Расположение мотивов на последовательностях (Шайн-Дальгарно голубой): 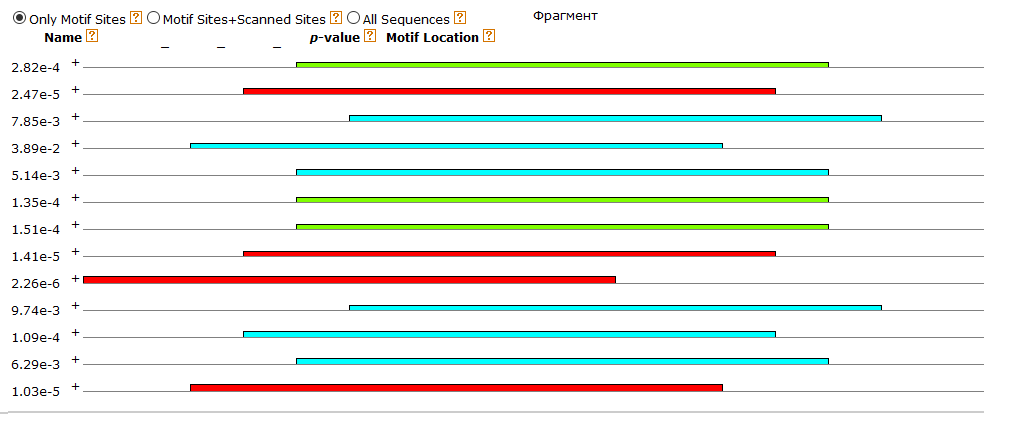 Лого трёх найденных мотивов:  По стрелке "вправо" рядом с каждым мотивом его можно передать в сервис FIMO, который найдет частоты встречаемости мотива в предложенном ему fasta-файле с координатами 17-нуклеотидных промоторных участков всех (а не только отобранных) генов. Для загрузки файла надо выбрать в первом выпадающем списке раздела "Input the sequences" строку "Upload sequences". Если p-value будет слишком малым, то в выдаче может не оказаться ничего, либо только один ген, как получилось у меня при p-value = е-06. Выдача FIMO для p-value = e-05: 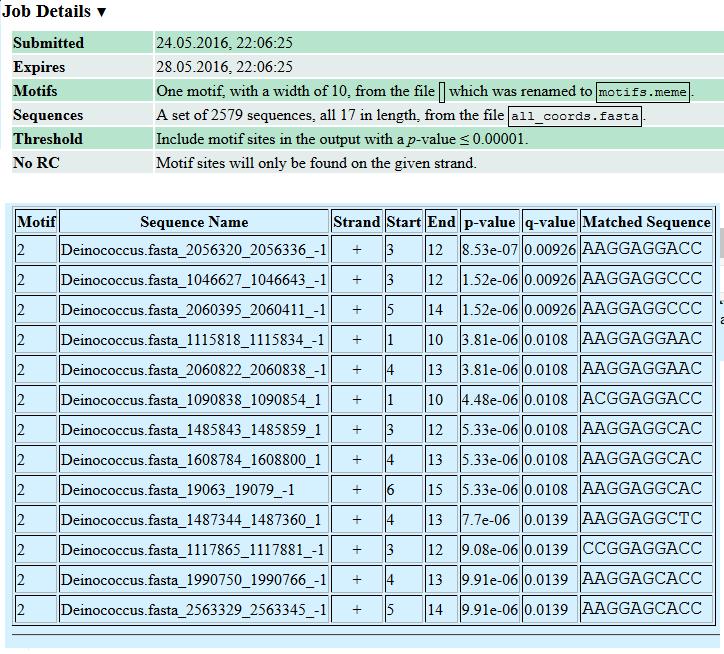 При p-value = e-05 FIMO выдал 13 генов с мотивом Шайн-Дальгарно, а повысив ещё на порядок, получим 88. Наконец, при p-value < 0.01 получим 604 гена, что составляет больше пятой части генов первой хромосомы Deinococcus radiodurans. Все результаты доступны в файле Deinococcus.xls Задание 3Для картирования была взята последовательность №18, скопирована и переименована в my18. Анализ качества прочтений показал, что триммирование необязательно:  bwa вывел информацию о 9256 прочтениях. После обработки samtools (Utilities for the Sequence Alignment/Map (SAM) format, команды и их описание можно найти здесь:Семестр3, Ресеквенирование) данные из idxstats были выведены в файл, содержимое которого после обработки в excel и удаления последовательностей, куда read'ы не откартировались вовсе, выглядит так: 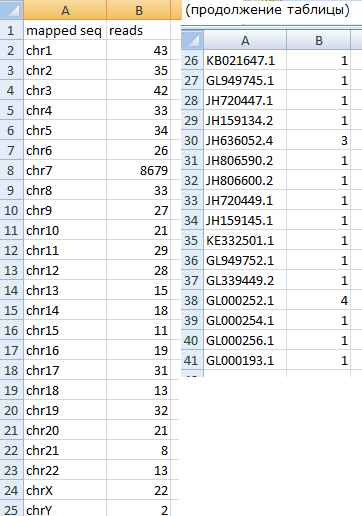 Наибольшее число прочтений пришлось на 7-ую хромосому. Model-based Analysis for ChIP-Sequencing ( macs2 callpeak -t chipseq_chunkX.sorted.bam) сообщила, что пиков слишком мало, поэтому было перезапущено с опцией --nomodel. Получены my18_peaks.xls и my18_peaks.narrowPeak с результатами macs.Для наглядного отображения в геномный браузер UCSC был загружен my18_peaks.narrowPeak c добавленой вверху строчкой описания данных: track type=narrowPeak visibility=3 db=hg19 name="my18" и выбрана сборка генома hg19 в выпадающем списке сверху. После перехода в геномном браузере на 7ую хромосому, нуклеотиды 148611200-149416700, пики можно видеть на cкриншоте (рыжими чёрточками): . Описание пиков:Таблица с характеристиками пиков (месторасположение, максимум, смещение максимума и т.п.): 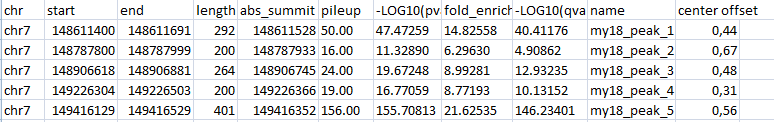 Наиболее достоверными по отрицательным логарифмам p-value и q-value являются пики 1 и 5. Ширина пиков от 200 до 400, хуже всех центрированы пики 2 и 4.
Пик 1 не попадает в какую-либо известную значимую область, если только не считать эту область промоторной для гена PDIA4 (protein disulfide isomerase family A) (на скриншоте не показана, по текущей аннотации начинается с 148700154):
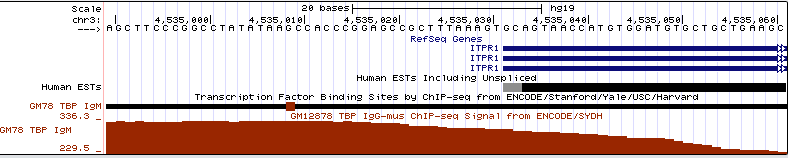
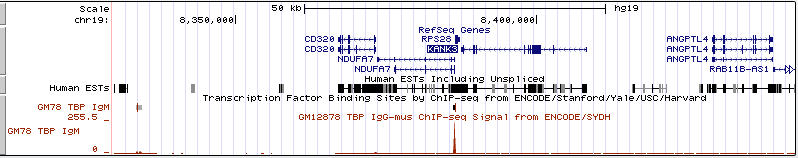
Задание 4.Для клеточной линии HCT116 Поиск TATA-бокса по браузеру оказался довольно утомительным: из 15 просмотренных пиков в промоторных областях генов только 2 таких области содержали его. Из этого можно сделать вывод, что для человека TATA-бокс нетипичен. Содержащие ТАТА-бокс промотеры:
Обзорный вид: 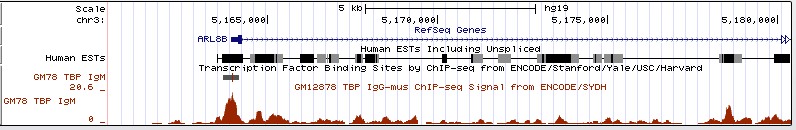 В масштабе нуклеотидов: 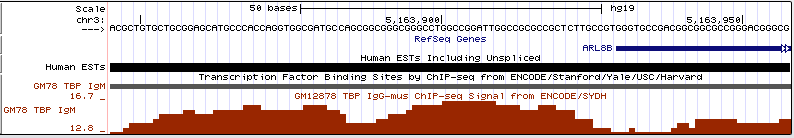 |