Эволюционные домены
Выбор домена
Для данной работы выбрала MipZ - АТФаза, образующая комплекс с белком ParB, разделяющим хромосомы вблизи ориджина репликации. Он отвечает за временную и пространственную регуляцию образования FtsZ кольца. Pfam AC PF09140, Pfam ID MipZ. Домен входит в 431 последовательность из 400 организмов, входит в 7 доменных архитектур, в базе данных PDB находится 5 трехмерных структур. Ниже приведено изображение каталитической субъединицы, содержащий данный домен (рис. 1). Страница домена в Pfam.
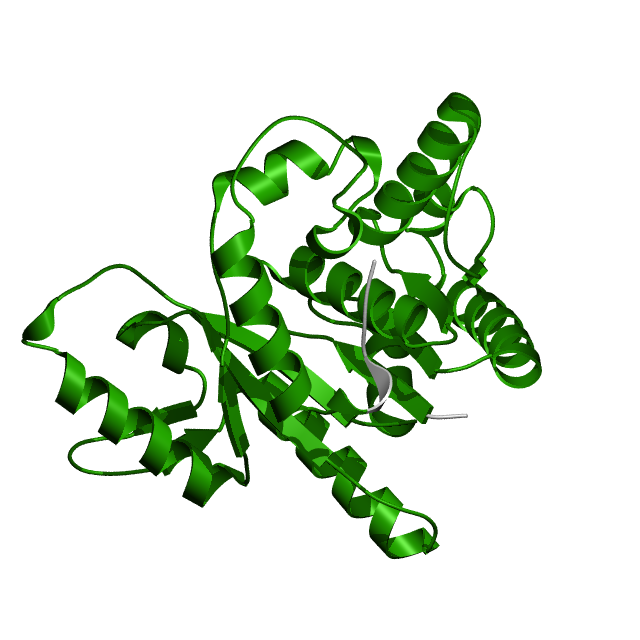
Рисунок 1.АТФаза MipZ, пространственная структура.
С помощью Jalview загрузила выравнивание домена. Ассоциировала структуру 2XJ9 c Q9A6C9_CAUCR. Сделала раскраску по ClustalX и консервативность 13%. Сохранила выравнивание в формате .fasta и .jar.
Выбрала три доменные архитектуры, которые показались мне наиболее интересными, для дальнейшего изучения - белок с одним доменом MipZ (PF09140); DUF59 (PF01883) + MipZ (PF09140) + ParA (PF10609); MipZ (PF09140) + ParA (PF10609). Это наиболее "популярные" доменные архитектуры, их 175, 127 и 118 последовательностей соответственно. PF10609 - ParA/MinD ATФаза-подобный домен, DUF59 (PF01883) - домен с неизвестной функцией, находится в цитоплазме многих организмов. Остальных доменных архитектур всего 4, а последовательностей из них 11 штук, поэтому описывать их не будем. Ссылка на список архитектур в Pfam.



Получила таблицу с информацией об архитектуре всех последовательностей, содержащих выбранный домен с помощью скрипта swisspfam_to_xls.py. Составила список последовательностей с указанием доменной архитектуры. Использовала сводную таблицу в Excel: строки – AC последовательностей, столбцы – домены Pfam. Скачала полные записи всех последовательностей из Uniprot, запустила скрипт uniprot_to_taxonomy.py. Перенесла таксономию с основную таблицу. Далее в качестве таксона выбрала все клеточные организмы, а в качестве его подтаксонов - Archaea и Bacteria.
Выбрала по 25 последовательностей из каждой выбранной выше архитектуры. С помощью скрипта filter_alignment.py оставила только выбранные последовательности из выравнивания. Далее открыла проект в JalView, удалила пустые колонки, создала группы по архитектурам (сверху находятся последовательности, взятые из белков с одним доменом, внизу - последовательности в архитектуре), удалила плохо выровненные участки. Сохранила выравнивание в формате .jar. Ниже привожу изображение выравнивания (ClustalX, консервативноcть 30) (рис. 2). Участки, соответствующие альфа-спиралям и бета-тяжам, определены примерно, потому что выравнивание после удаления плохо выровненных участков значительно уменьшилось в длине, и остались только намеки на вторичные структуры.
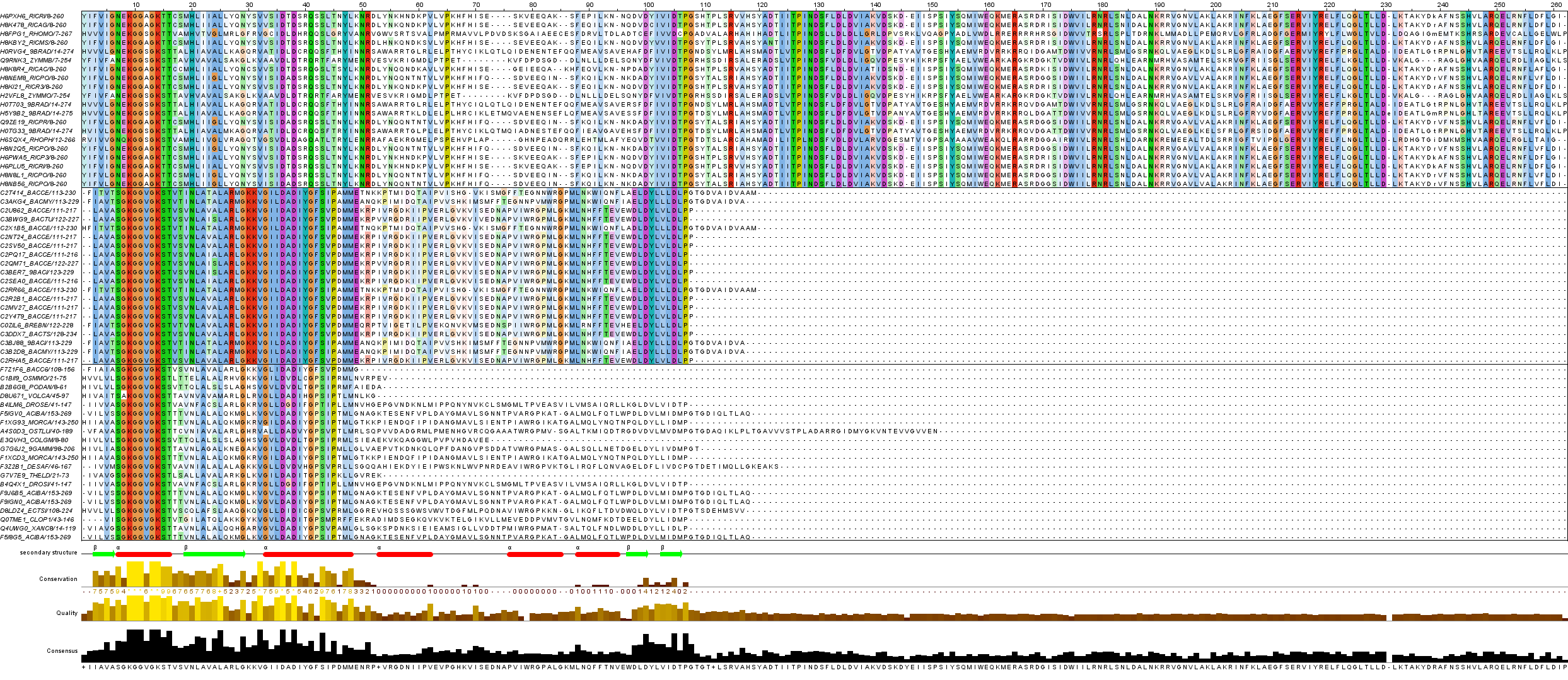
Рисунок 2.Выравнивание отобранных последовательностей домена, сверху группа c одним доменом MipZ (PF09140); далее DUF59 (PF01883) + MipZ (PF09140) + ParA (PF10609); MipZ (PF09140) + ParA (PF10609).
Исходя из выравнивания, все-таки можно понять, что большая консервативность в значимых участках - альфа-спиралях и бета-тяжах - существует. Наибольшая консервативность наблюдается у трехдоменных белков. Для дальшейших исследований будем смотреть на одно- и трехдоменные архитектуры, так как в ходе улучшения выравнивания их выборка оказалась наиболее удачной.