Нахождение и описание полиморфизмов у пациента
Мне были даны одноконцевые чтения экзома из хромосомы №7 пациента. Я скачала файл с этими чтениями chr7.fastq и файл с референсной последовательностью хромосомы chr7.fasta
1. Анализ качества чтений.
Чтобы проанализировать качество чтений, я применила программу FastQC. Я запустила следующую команду:
fastqc chr7.fastqЯ получила 2 файла: chr7_fastqc.html и chr7_fastqc.zip. Первый файл - это анализ качества последовательностей в виде html страницы, а второй - архив со служебными файлами для html страницы.
2. Очистка чтений
Так как адаптеры в чтениях уже удалены, мне нужно было всего лишь отрезать с конца каждого чтения нуклеотиды с качеством ниже 20 и оставить только чтения длиной не меньше 50 нуклеотидов. Я делала очистку с помощью программы Trimmomatic. Я запустила такую команду:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr7.fastq clear.fastq SLIDINGWINDOW:3:20 LEADING:20 TRAILING:20 MINLEN:50 fastqc clear.fastqПервая команда была направлена на очистку чтений от низкокачественных концов. Для этого применялась операция SLIDINGWINDOW, которая прошлась по прочтениям окном длиной 3 и удалила правый конец каждого прочтения после окна со средним качеством меньше 20; операция LEADING, удаляющая с начала каждого рида нуклеотиды качеством ниже 20 (хотя таких и не было, я включила эту операцию просто для примера); TRAILING, удаляющая некачественные нуклеотиды с конца рида (это должна была сделать SLIDINGWINDOW, но TRAILING я включила для подстраховки); MINLEN, удаляющая риды длины менее 50. Результат был записан в файл clear.fastq. Вторая команда была выполнена для анализа качества чтений. Результат: clear_fastqc.html
Если в файле до чистки было 3752 рида, то после чистки их осталось 3313. Длина ридов до чистки лежала в диапазоне 30-100, а после чистки - в диапазоне 50-100. На рис.1 сравнение качества ридов по парам оснований до и после чистки.
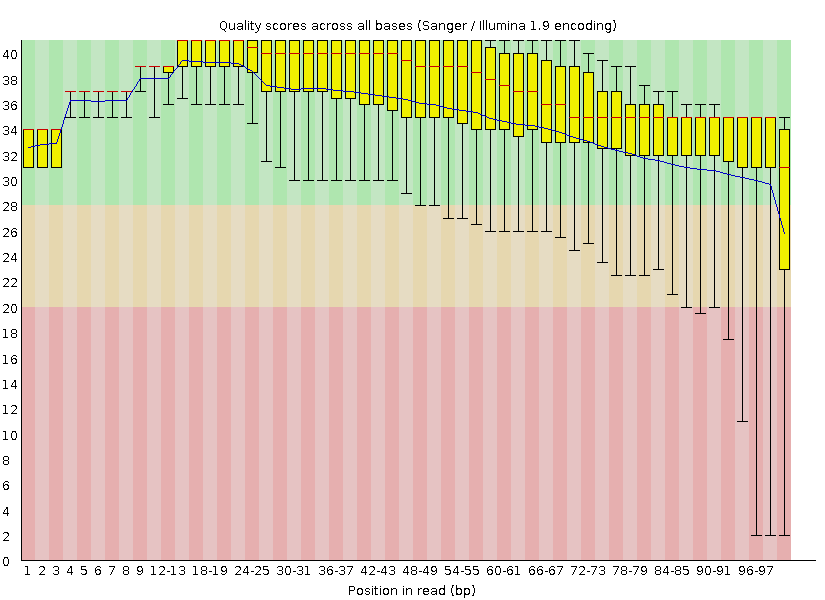 Рис.1а. Качество ридов по парам оснований до очистки. |
Рис.1б. Качество ридов по парам оснований после очистки. |
Видно, что хотя некачественные нуклеотиды удалялись только с концов, качество ридов улучшилось на всем протяжении цепей. К тому же, из рис.1а видно, что некачественные участки, которые должны были быть отрезаны при чистке, составляют не более 10 нуклеотидов. Поэтому можно сделать вывод, что, во-первых, риды с некачественными концами были не очень качественными на всем протяжении, а во вторых, самые некачественные риды были довольно короткими (если учесть то, что верхняя граница длин ридов не изменилась, несмотря на очистку).
На рис.2 представлено распределение ридов по качеству. Видно, что после чистки исчезли риды качества ниже 28, а ридов качества ниже 33 стало меньше.
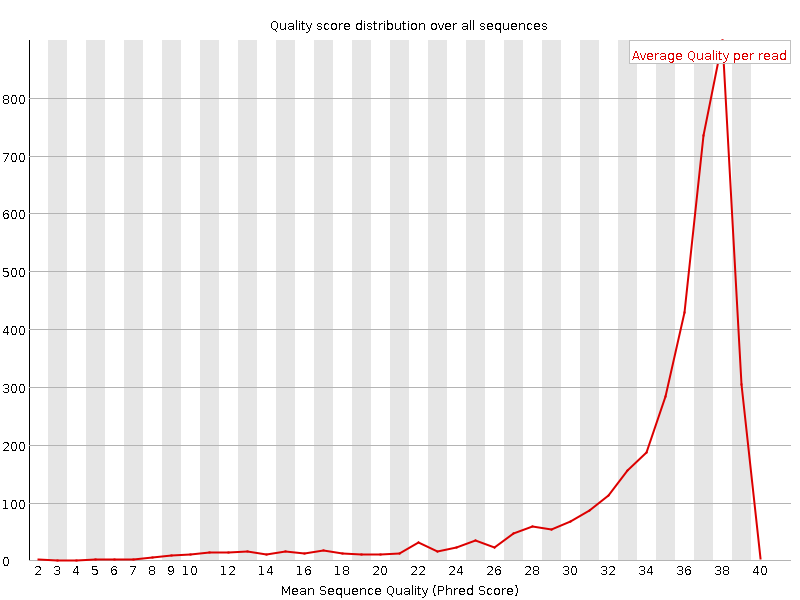 Рис.2а. Распределение ридов по качеству до очистки. |
Рис.2б. Распределение ридов по качеству после очистки. |
Примечание: на самом деле команда, которая использовалась для очистки прочтений, не вполне подходит для поставленной задачи. Мне следовало использовать такую команду:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr7.fastq clear1.fastq TRAILING:20 MINLEN:50 fastqc clear1.fastqПолучилось так: clear1_fastqc.html. Видно, что качество ридов удовлетворительное, и при этом количество оставшихся ридов больше - 3650. Несмотря на это, вся остальная работа выполнена для файла clear.fastq, потому что ошибка была найдена слишком поздно.
3. Картирование чтений
Для картирования чтений использовалась программа BWA. Сначала я проиндексировала референсную последовательность командой:
bwa index chr7.fastaЭта команда создала несколько файлов форматов amb, ann, bwt, pac, sa. Затем я выровнила мои риды с референсом алгоритмом mem командой:
bwa mem chr7.fasta clear.fastq > cart.samПолучился файл cart.sam с картированными ридами.
4. Анализ выравнивания
Для перевода файла sam в файл bam использовалась команда:
samtools view cart.sam -b -o cart.bamДалее полученный файл cart.bam был отсортирован по координатам:
samtools sort cart.bam -o sort.bamЭта команда выдала промежуточные файлы в stdout и зависла. Поэтому я использовала такую команду:
samtools sort cart.bam -T prefix -o sort.bam(Эта команда делала то же самое, но промежуточные файлы вместо stdout записала в отдельный файл prefix.nnnn.bam, который потом удалился.) Получился файл sort.bam, который я проиндексировала командой:
samtools index sort.bamЭта команда создала файл sort.bam bai. Чтобы выяснить, сколько чтений откартировалось на геном, я сделала так:
samtools idxstats sort.bamВыдача:
chr7 159138663 3313 0 * 0 0 0В каждой строчке записаны по порядку: название референса, длина последовательности, кол-во откартированных ридов, кол-во неоткартированных ридов. Как мы видим, откартировались все риды. Чтобы узнать покрытие ридами каждого нуклеотида, я запустила команду:
samtools depth sort.bam > depth.outПолучился файл depth.out. На рис.3 гистограмма с покрытиями референса.
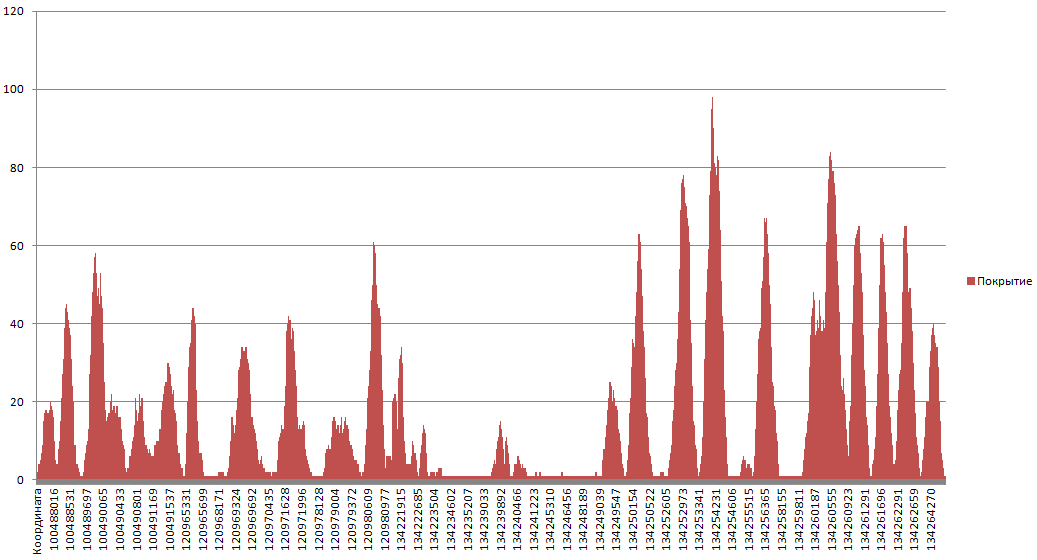
Рис.3. Покрытие референса ридами.
Как мы видим, самый покрытый ридами нуклеотид имеет координату 134254165. Я определила его местонахождение в референсном геноме Homo sapiens chromosome 7. Оказалось, что этот нуклеотид расположен в гене AKR1B15, из семейства альдо-кето редуктаз, причем в экзоне (координаты экзона 134254165-134254281). Чтобы определить покрытие экзона, я сделала так:
samtools depth sort.bam -r chr7:134254165-134254281 > depth_exon.outПолучился файл depth_exon.out. На рис.4 гистограмма с покрытиями экзона. Среднее покрытие экзона 80.675 (среднее арифметическое покрытий всех позиций в экзоне).
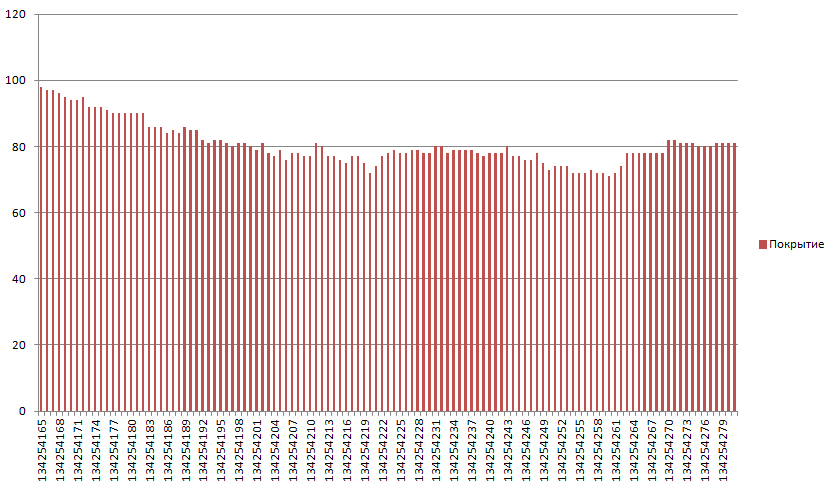
Рис.4. Покрытие экзона ридами.
5. Поиск SNP и инделей.
Чтобы получить список полиморфизмов в бинарном формате, использовалась команда:
samtools mpileup sort.bam -u -f chr7.fasta > polymorph.bcfПолучился файл polymorph.bcf. Для получения из него списка отличий между референсом и чтениями я сделала так:
bcftools call polymorph.bcf -cv -o polymorph.vcfПолучился файл polymorph.vcf. В нем 31 полиморфизм, из них 3 - индели, а остальные - замены. Вот для примера 3 полиморфизма:
| Координата | Полиморфизм | В референсе | В ридах | Покрытие | Качество | Изображение IGV |
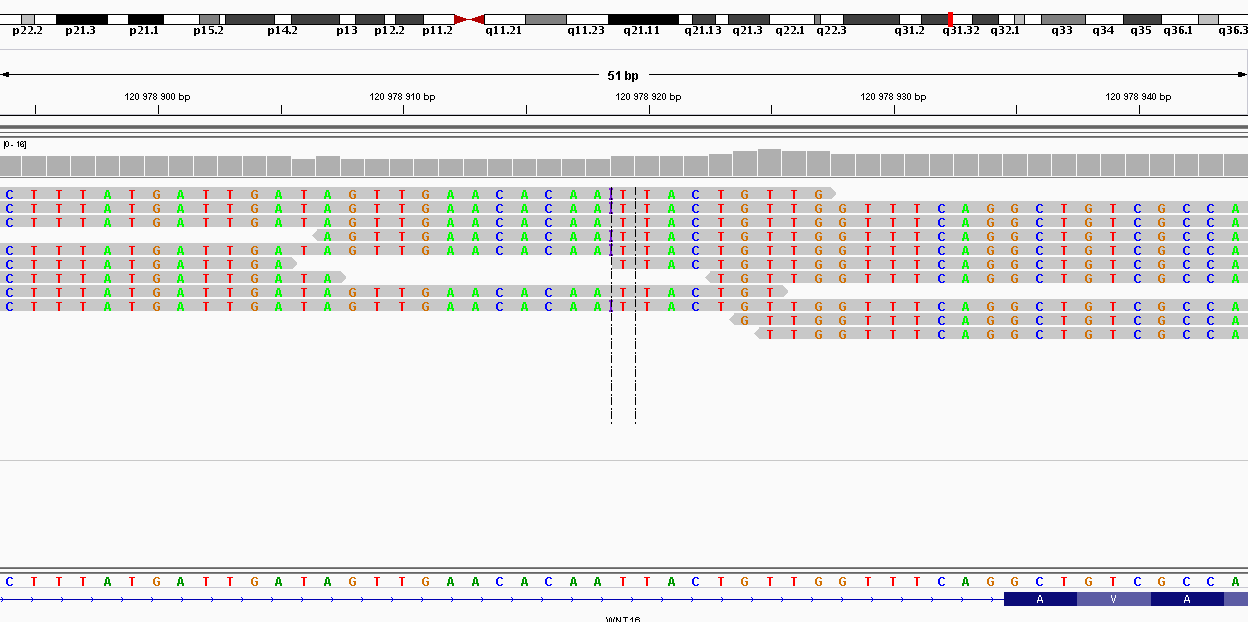
| 120978918 | Индель | ATT | ATTT | 7 | 107.467 | Индель |
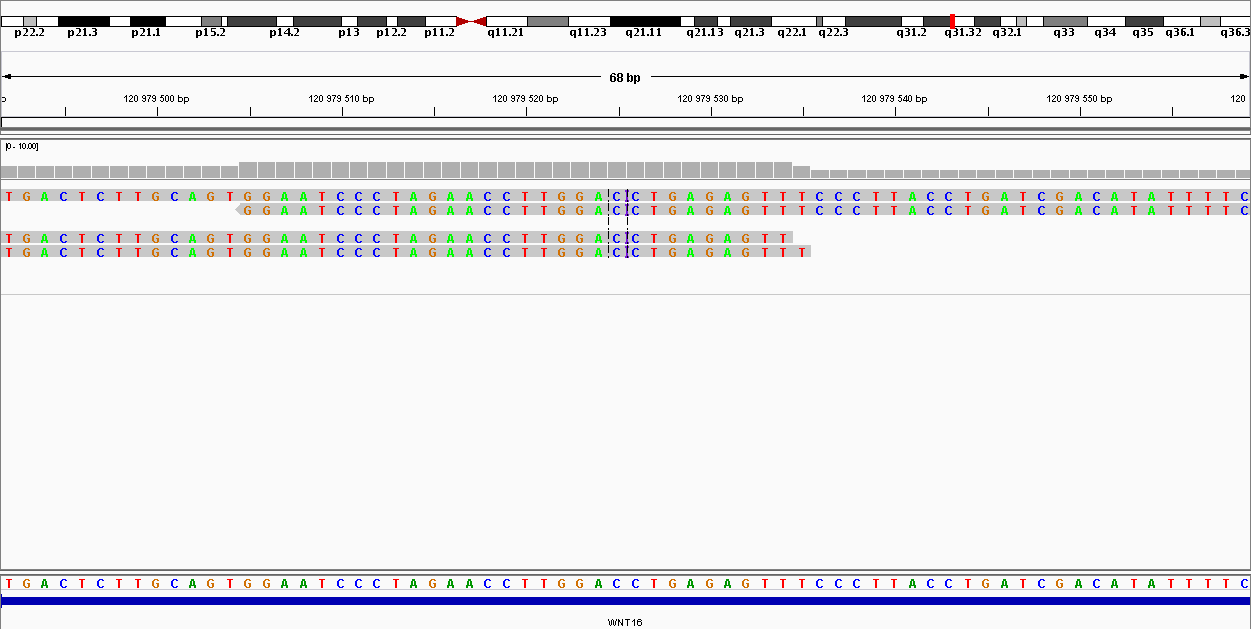
| 120979525 | Индель | CCT | CCTCTCT | 4 | 143.973 | Индель |
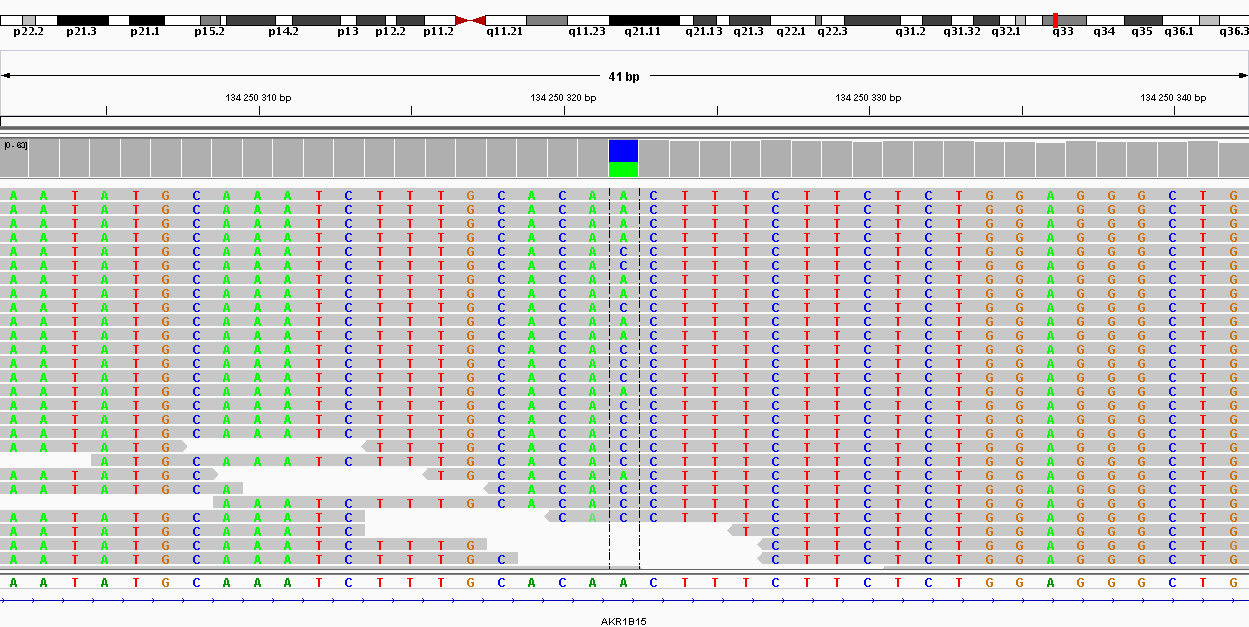
| 134250322 | Замена | A | C | 61 | 225.009 | SNP |
{kind=link}
{kind=link}
{kind=link}
6. Аннотация SNP.
Для аннотации использовалась программа annovar. Для перевода файла в формат, совместимый с ней я запустила команду:
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 polymorph.vcf > polymorph.avinputПолучился файл polymorph.avinput. Далее я проаннотировала найденные полиморфизмы в пяти базах данных. Для этого я создала 5 директорий для файлов с аннотациями из соотв. баз данных: refgene, dbsnp, 1000_genomes, gwas, clinvar. А потом проаннотировала, используя программу annotate_variation.pl. (Индели тоже были проаннотированы)
1. dbsnp
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out dbsnp/out -build hg19 -dbtype snp138 polymorph.avinput /nfs/srv/databases/annovar/humandb/В результате получилось 3 файла: out.log, out.hg19_snp138.dropped, out.hg19_snp138.filtered. В первом всякая информация о процессе работы, во втором snp и индели, содержащие rs (т.е. аннотированные в dbsnp) - их было 25 snp и 1 индель, а в третьем не содержащие - 3 snp и 2 инделя. Гомозиготных полиморфизмов 10, а гетерозиготных - 21.
2. refgene
perl /nfs/srv/databases/annovar/annotate_variation.pl -out refgene/out -build hg19 polymorph.avinput /nfs/srv/databases/annovar/humandb/Было получено опять 3 файла: out.log (опять с информацией о процессе), out.variant_function (с информацией о всех полиморфизмах) и out.exonic_variant_function (с информацией о полиморфизмах, попавших в экзоны). Итог: в экзоны попало 4 snp, в UTR3 - 2 snp и 1 индель, в интроны - 22 snp и 2 инделя. Вот таблица с экзонными snp:
| Координата | Тип замены | Ген | Нуклеотидная замена | Аминокислотная замена | Глубина покрытия | Качество ридов | Изображение в IGV |
| 100490077 | synonymous | ACHE | C1431T | P477P | 28 | 221.999 | SNP |
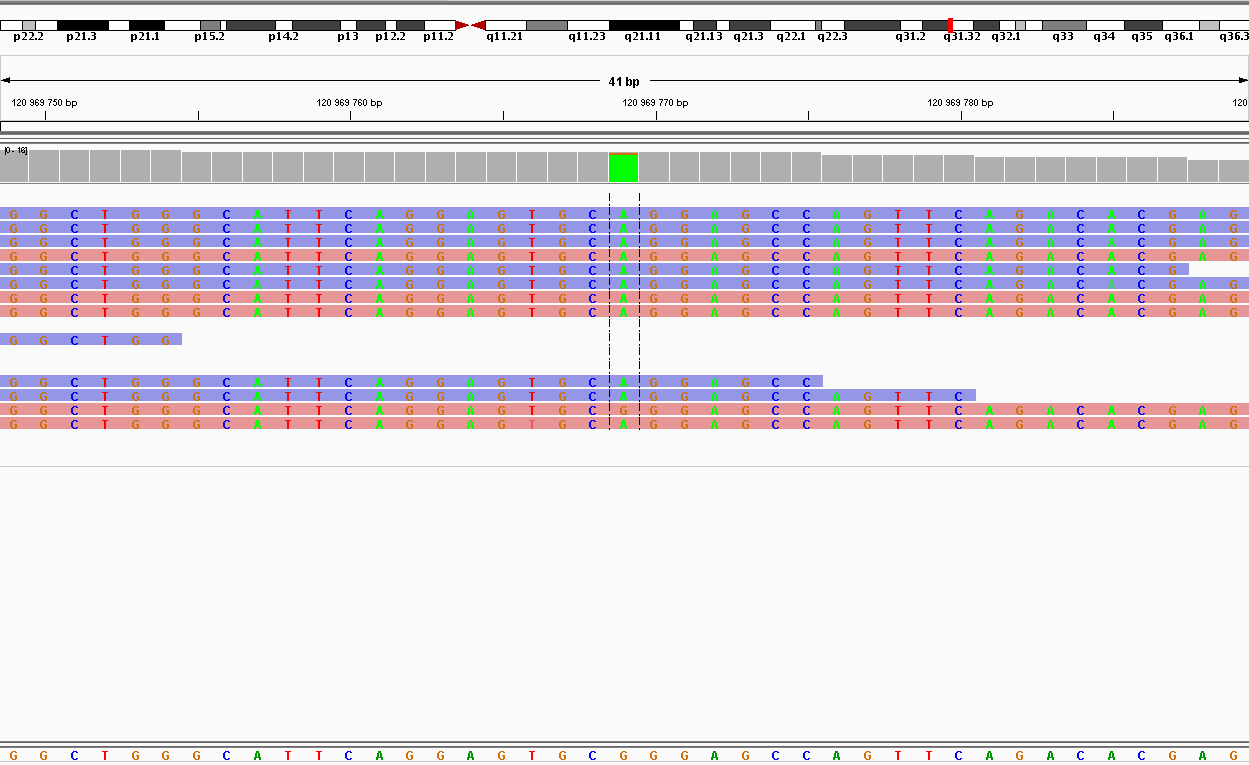
| 120969769 | nonsynonymous | WNT16 | G214A | G72R | 12 | 209.014 | SNP |
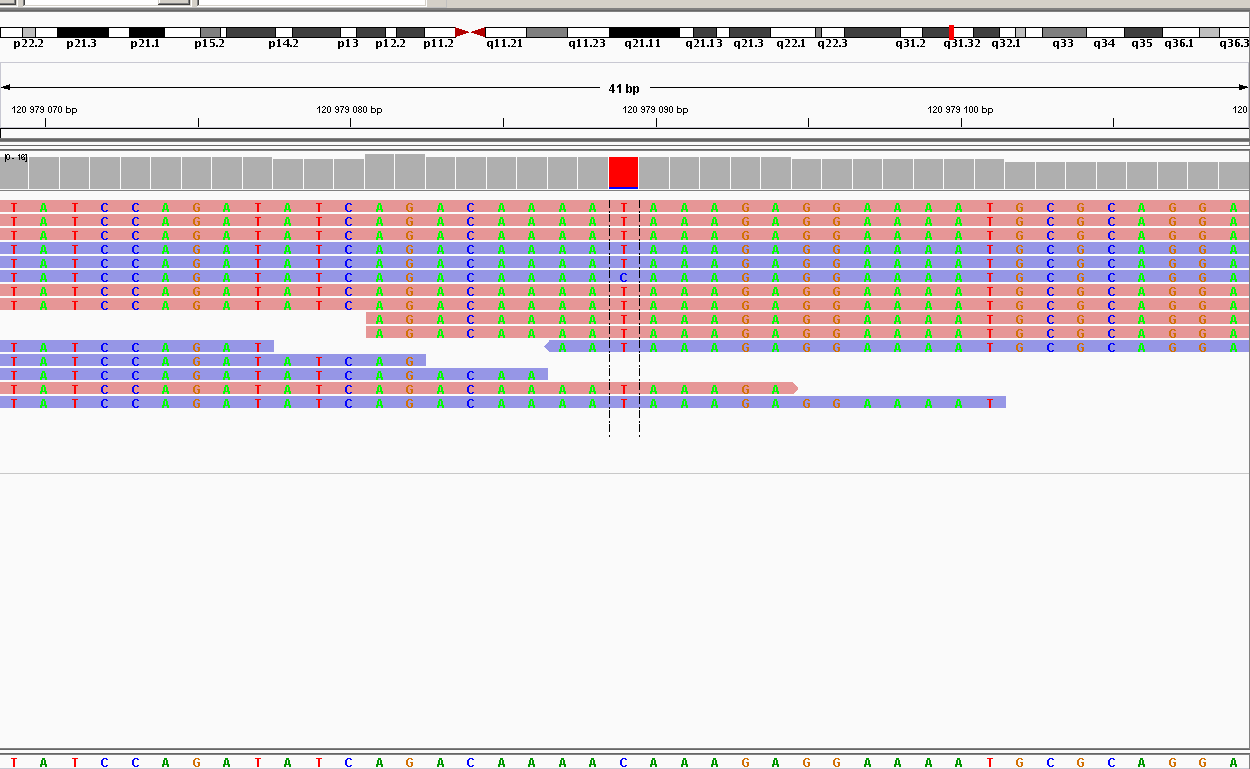
| 120979089 | nonsynonymous | WNT16 | C758T | T253I | 13 | 225.2 | SNP |
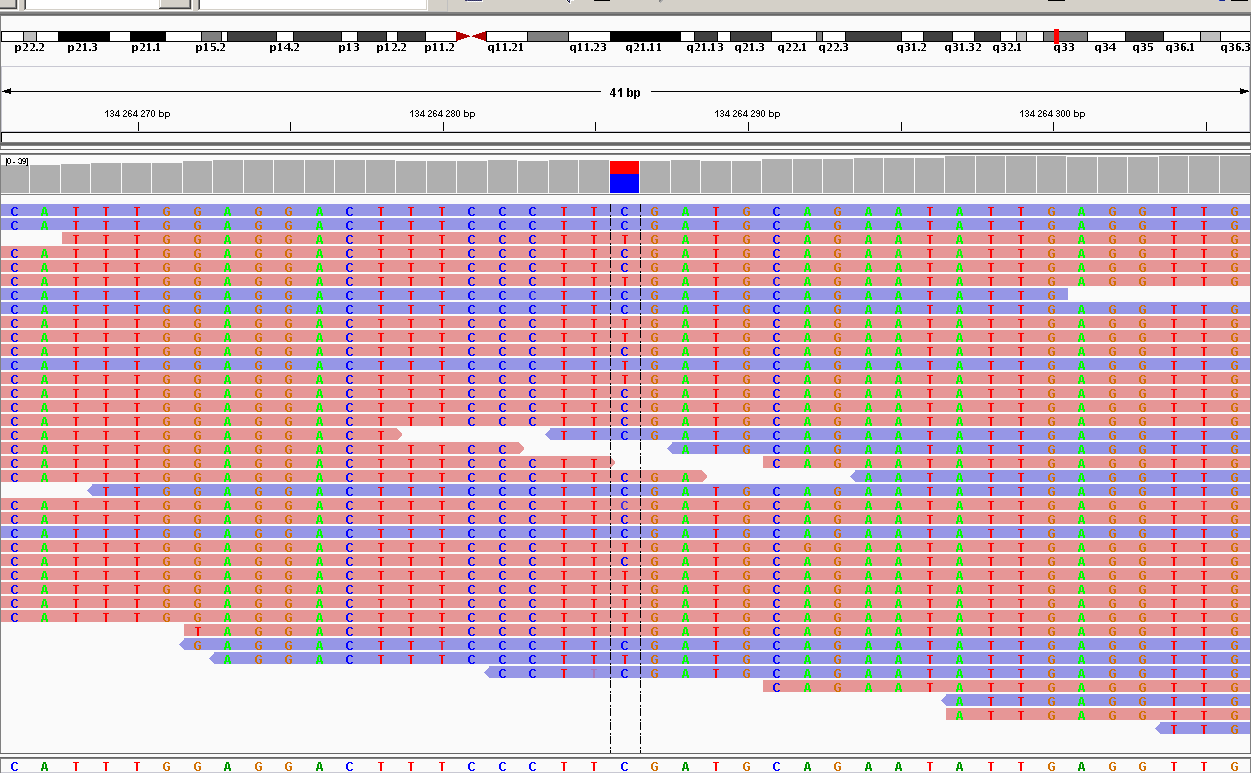
| 134264286 | synonymous | AKR1B15 | C1020T | F340F | 32 | 188.009 | SNP |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Как мы видим, 2 замены оказались синонимичными и попали в гены ацетилхолинэстеразы и альдокеторедуктазы, они не влияют на трансляцию белка. Но две несинонимичные замены оказались в одном гене WNT16, который принимает участие в эмбриогенезе, регуляции клеточной дифференциации и других процессах. Можно предположить, что этот ген был поврежден.
3. 1000 genomes
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out dbsnp/out -build hg19 -dbtype snp138 polymorph.avinput /nfs/srv/databases/annovar/humandb/Как и в dbsnp, получилось 3 файла: out.log, out.hg19_ALL.sites.2014_10_dropped, out.hg19_ALL.sites.2014_10_filtered. В первом всякая информация о процессе работы, во втором snp и индели, аннотированные в базе - 25 snp, а в третьем не аннотированные - 3 snp и 3 инделя. К каждому аннотированному полиморфизму прилагалась частота встречаемости этого полиморфизма у людей. В моем случае она варьировала от 0.0239617 до 0.510383. Примечательно, что все экзонные замены, аннотированные refgene, были аннотированы и здесь, и встречались достаточно часто (>0.1).
4. Gwas
perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -build hg19 -out gwas/out -dbtype gwasCatalog polymorph.avinput /nfs/srv/databases/annovar/humandb/Было получено 2 файла: out.gwas.log и out.gwas.hg19_gwasCatalog (содержит snp, для которых известно какое-то клиническое значение). Во второй файл попало 4 snp. Три из них совпали с экзонными snp, аннотированными refgene: синонимичная замена из гена ACHE вызывает предрасположенность к диабету 2 типа, 2 несинонимичных из гена WNT16 влияют соответственно на минеральную плотность костной ткани (Bone mineral density) и толщину коркового слоя кости (Cortical thickness). Последняя замена влияет на продолжительность жизни.
5. Clinvar
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out clinvar/out -buildver hg19 -dbtype clinvar_20150629 polymorph.avinput /nfs/srv/databases/annovar/humandb/И снова получилось 3 файла: out.log, out.hg19_clinvar_20150629_filtered, out.hg19_clinvar_20150629_dropped. В файл dropped ничего не попало, т.е. аннотированных в clinvar полиморфизмов не оказалось.
Вот таблица со всеми полиморфизмами и их аннотациями в разных б.д. Polymorph.xlsx. А на рис.5 изображение выравнивания в IGV.
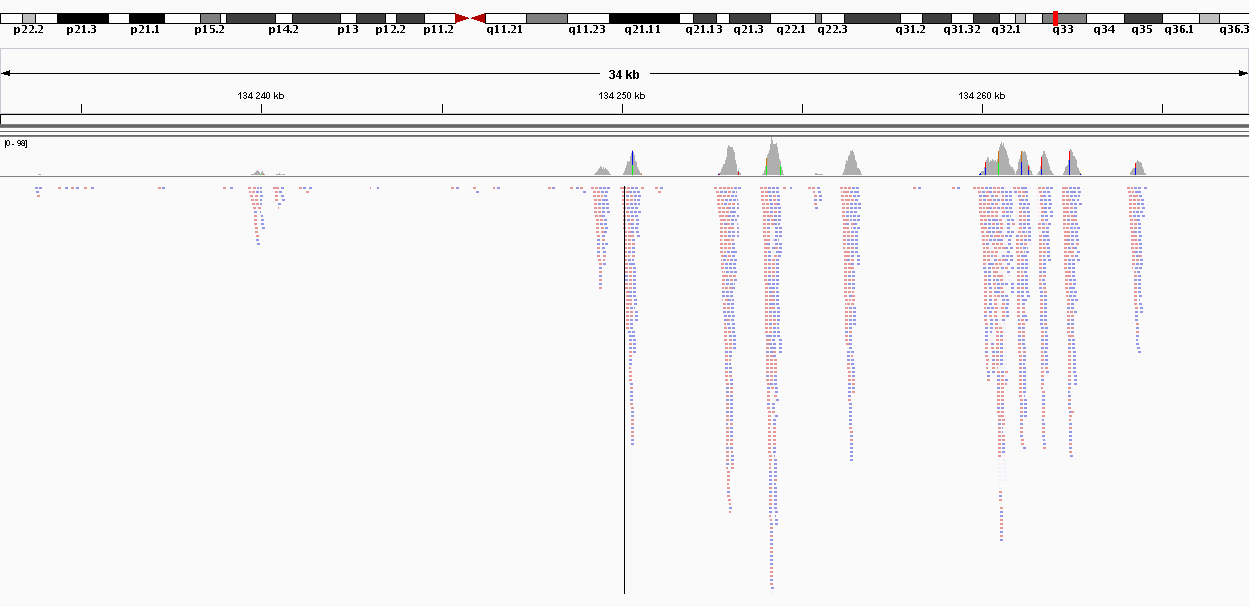
Рис.5. Изображение выравнивания в IGV.