Занятие 4: Реконструкция деревьев по нуклеотидным последовательностям. Анализ деревьев, содержащих паралоги
Построение дерева по нуклеотидным последовательностям
Для отобранных на предыдущем занятии бактерий требуется построить филогенетическое дерево, используя последовательности РНК малой субъединицы рибосомы (16S rRNA). Для этого сначала необходимо найти последовательности 16S рибосомальной РНК каждой из бактерий. Это можно сделать следующим способом. В записи EMBL, описывающей полный геном бактерии, найти соответствующее поле (FT с FTkey rRNA и упоминанием 16S rRNA в примечании). Затем вырезать нужный участок из записи EMBL с использованием программы seqret.
AC записей EMBL и координаты (одной из) 16S rRNA для отобранных бактерий
| Название | Мнемоника | AC записи EMBL | Начало | Конец | Последовательность (+/-) |
| Bacillus subtilis | BACSU | AL009126 | 30279 | 31832 | + |
| Clostridium tetani | CLOTE | AE015927 | 176113 | 177621 | + |
| Enterococcus faecalis | ENTFA | AE016830 | 248466 | 249987 | + |
| Geobacillus kaustophilus | GEOKA | BA000043 | 10421 | 11973 | + |
| Lactobacillus delbrueckii | LACDA | CR954253 | 45160 | 46720 | + |
| Listeria monocytogenes | LISMO | AL591974 | 37466 | 39020 | + |
| Staphylococcus epidermidis | STAES | AE015929 | 1598006 | 1599559 | - |
Полученные описанным выше образом последовательности были сохранены в соответствующем файле, при этом названия последовательностей были отредактированы так, чтобы они отвечали мнемонике организмов. Выравнивание последовательностей было получено с помощью программы JalView (Web Service > Alignment > Muscle with Defaults).
Затем выравнивание было импортировано в программу MEGA (как выравнивание нуклеотидных последовательностей), с помощью которой было построено дерево методом Neighbor-Joining. Полученное дерево приведено на изображении ниже.
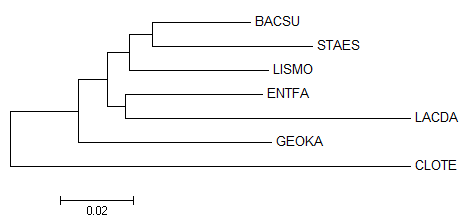
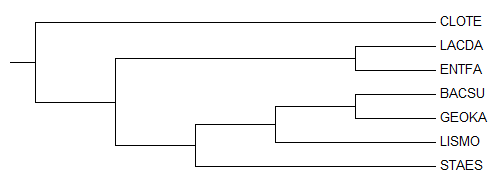
Как видно из сравнения с правильным деревом (приведено на изображении ниже), полученное дерево имеет с ним лишь одну общую нетривиальную ветвь ({LACDA, ENTFA} vs {CLOTE, BACSU, GEOKA, LISMO, STAES}).
Попытаться "улучшить" дерево можно, например, с помощью применения бутстрэп-анализа. Ещё один способ — удалить из выравнивания, которое является основой для построения дерева, концевые участки, которые присутствуют не во всех последовательностях и тем самым могут "портить" результат (если таковые вообще имеются). В данном случае было удалено 10 и 35 нуклеотидов с начала и конца выравнивания соответственно. Ни одна из попыток не привела к "улучшению" дерева (то есть его приближению к правильному, например, по количеству общих нетривиальных ветвей).
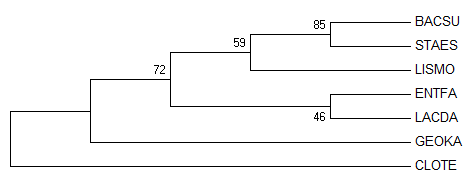
Таким образом, качество реконструкции дерева по последовательности РНК малой субъединицы рибосомы оказалось ниже по сравнению с деревьями, реконструированными по белкам.
Построение и анализ дерева, содержащего паралоги
Для гомологов белка CLPX_BACSU в отобранных бактериях необходимо построить дерево.
Для поиска гомологов предложен файл proteo.fasta, содержащий записи банка Uniprot, относящиеся к исходному списку бактерий. Поиск гомологов можно произвести с помощью программы blastp, а затем отобрать находки, относящиеся к отобранным бактериям. Последовательность команд для этого приведена ниже.
seqret sw:clpx_bacsu
makeblastdb -in /P/y11/Term_4/proteo.fasta -out proteo -dbtype prot
blastp -query clpx_bacsu.fasta -db proteo -evalue 0.001 -out clpx_blastp.out -outfmt 6
for id in bacsu clote entfa geoka lacda lismo staes; do awk '{ print $2 }' clpx_blastp.out | grep -i $id >> clpx_homologs_ids.txt; done
В файле clpx_homologs_ids.txt получены идентификаторы гомологов белка CLPX_BACSU в отобранных бактериях в том виде, как они приведены в исходном файле proteo.fasta. Т.к. blastp не предоставляет подходящего для наших целей (получение множественного выравнивания) способа отображения результатов работы, то можно поступить следующим образом: с помощью скрипта (использован BioPython) "вытащим" последовательности, соответствующие полученным идентификаторам, из исходного файла proteo.fasta. Привести имена последовательностей в полученном файле clpx_homologs_seqs.fasta к форме белок_вид можно с помощью следующей команды:
sed -ri 's/>.*\|/>/' clpx_homologs_seqs.fasta
Построить выравнивание полученных последовательностей можно, например, с помощью программы muscle:
muscle -in clpx_homologs_seqs.fasta -out clpx_homologs_seqs_aligned.fasta
Полученный файл с выравниванием можно импортировать в программу MEGA, чтобы построить дерево. Ниже приведено дерево, реконструированное на основе выравнивания последовательностей гомологов белка CLPX_BACSU в отобранных бактериях методом Neighbor-Joining.

Если считать, что два гомологичных являются ортологами, если они из разных организмов и разделение их общего предка на линии, ведущие к ним, произошло в результате видообразования, то на построенном дереве ортологами являются, например, CLPX_LISMO и CLPX_BACSU, HSLU_LACDA и HSLU_ENTFA.
Если паралогами называть два гомологичных белка из одного организма, то на основе реконструированного дерева можно сказать, что паралогами являются, например, Q891B9_CLOTE и Q899H3_CLOTE, Q1G869_LACDA и Q1GBM8_LACDA.
Ссылки
- Файл 16s_rrna.fasta.
- Файл 16s_rrna_aligned.fasta.
- Файл 16s_rrna_aligned_notails.fasta.
- Файл clpx_bacsu.fasta.
- Файл clpx_homologs_ids.txt.
- Файл get_clpx_homologs.py.
- Файл clpx_homologs_seqs.fasta.
- Файл clpx_homologs_seqs_aligned.fasta.