Занятие 2: Реконструкция филогенетических деревьев
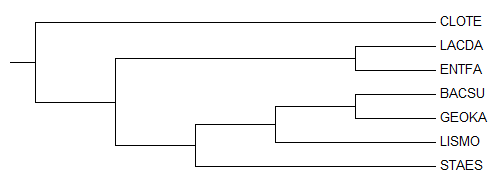
На предыдущем занятии были отобраны 7 бактерий, для которых было составлено филогенетическое дерево:
С помощью таксономического сервиса NCBI можно определить, к каким таксонам относятся выбранные бактерии:
| Название | Мнемоника | Тип | Класс | Отряд | Семейство |
| Bacillus subtilis | BACSU | Firmicutes | Bacilli | Bacillales | Bacillaceae |
| Clostridium tetani | CLOTE | Firmicutes | Clostridia | Clostridiales | Clostridiaceae |
| Enterococcus faecalis | ENTFA | Firmicutes | Bacilli | Lactobacillales | Enterococcaceae |
| Geobacillus kaustophilus | GEOKA | Firmicutes | Bacilli | Bacillales | Bacillaceae |
| Lactobacillus delbrueckii | LACDA | Firmicutes | Bacilli | Lactobacillales | Lactobacillaceae |
| Listeria monocytogenes | LISMO | Firmicutes | Bacilli | Bacillales | Listeriaceae |
| Staphylococcus epidermidis | STAES | Firmicutes | Bacilli | Bacillales | Staphylococcaceae |
На дереве отобранных бактерий можно найти ветви, выделяющие отдельные таксоны. Например, классы Bacilli и Clostridia разделены ветвью {CLOTE} vs {BACSU, ENTFA, GEOKA, LACDA, LISMO, STAES} (можно отметить, что эта ветвь не является нетривиальной). Отряд Lactobacillales выделен соответвующей ветвью (включающей два листа: ENTFA и LACDA); аналогичным образом выделен отряд Bacillales (ветвь {CLOTE, LACDA, ENTFA} vs {BACSU, GEOKA, LISMO, STAES}). Семество Bacillaceae также выделено ветвью: {BACSU, GEOKA} vs {CLOTE, LACDA, ENTFA, LISMO, STAES}.
Множественное выравнивание белков
Из предложенного списка функций белков была выбрана функция «Фактор инициации трансляции 2», которой соответствует мнемоника IF2. Получить из Swiss-Prot последовательности белков с данной функцией из отобранных бактерий можно с помощью программы JalView (File > Fetch Sequence(s)... и получить последовательности из Uniprot, введя через точку с запятой идентификаторы вида IF2_BACSU, где вторая часть идентификатора представляет собой мнемонику соответствующей бактерии) или аналогичным образом с использованием программы seqret:
seqret sw:if2_bacsu
Полученные последовательности были помещены в один файл в формате .fasta. При этом названия последовательностей были отредактированы так, чтобы они содержали только мнемонику видов.
Выравнивание отобранных белков можно получить, например, с помощью программы JalView (Web Service > Alignment > Muscle with Defaults). Ниже приведено изображение выравнивания в «блочной» форме с блоками шириной 100 остатков и раскраской по проценту идентичности.
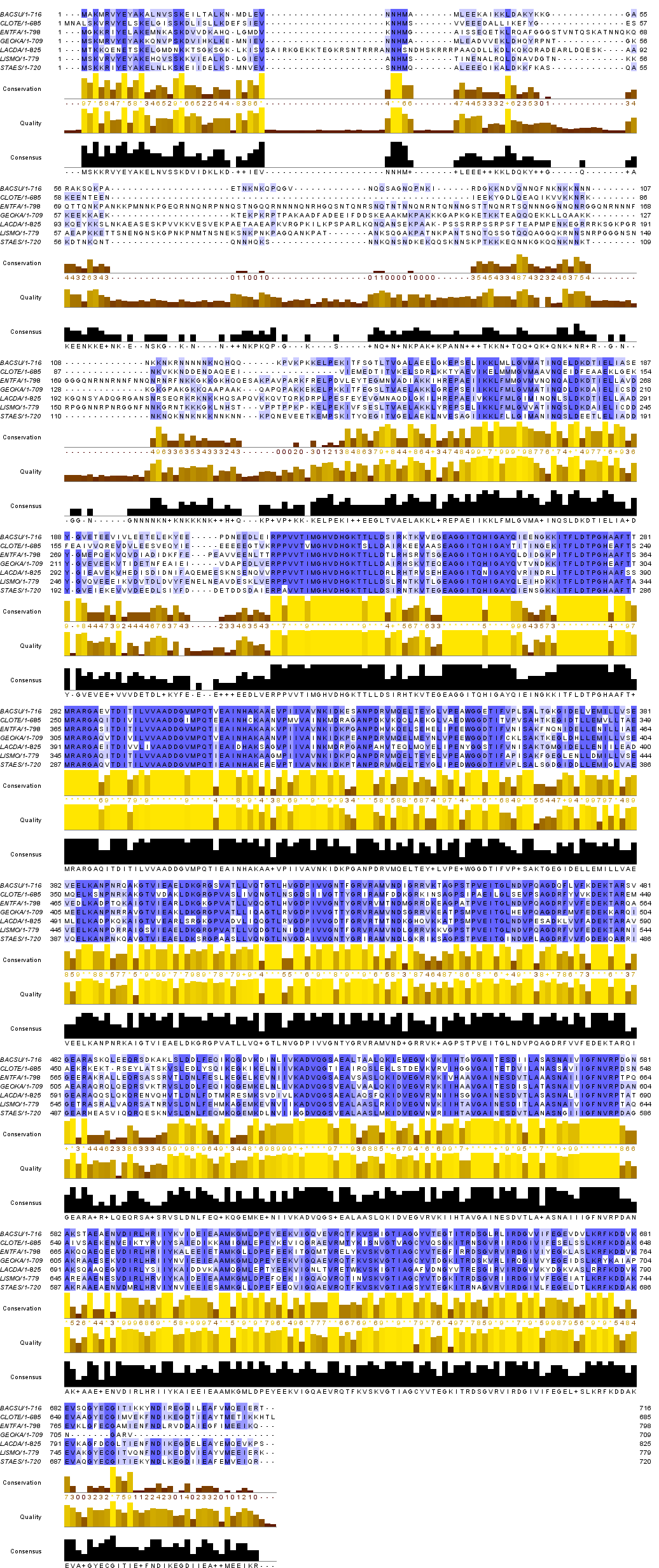
Построенное выравнивание отобранных белков, а также проект JalView были сохранены в соответствующих файлах.
В выравнивании можно найти несколько диагностических позиций, по которым можно судить о том, относится ли та или иная последовательность выравнивания к некоторому таксону. Например, на изображении ниже диагностические позиции, выделяющие класс Bacilli, отмечены красными указателями; выделяющие отряд Bacillales - зелёными.

Реконструкция филогенетического дерева
В JalView доступны четыре метода для реконструкции филогенетического дерева, которые доступны из меню Calculate > Calculate Tree. Каждое из деревьев было сохранено в соответствующем файле. Приведённые ниже изображения созданы с помощью программы Mega.
Average Distance Using % Identity
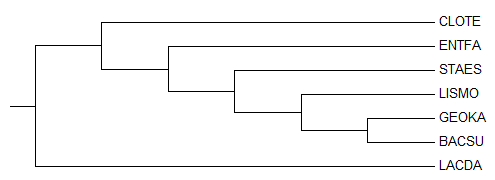
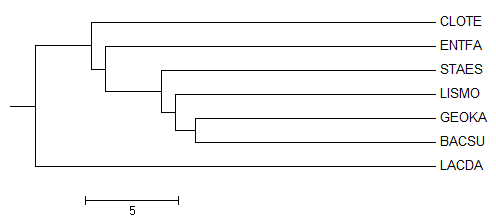
В этом дереве присутствует ветвь {CLOTE, LACDA} vs {ENTFA, STAES, LISMO, GEOKA, BACSU, LACDA}, которая отсутствует в правильном дереве, и отсутствует ветвь {LACDA, ENTFA} vs {CLOTE, BACSU, GEOKA, LISMO, STAES}, которая присутствует в правильном дереве.
Neighbour Joining Using % Identity
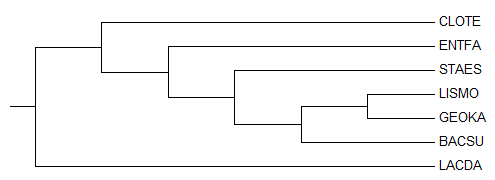
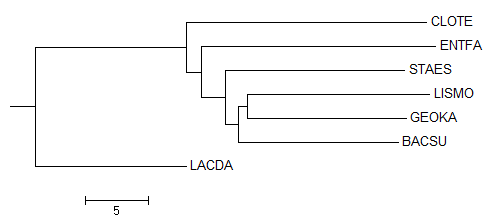
В этом дереве также присутствует ветвь {CLOTE, LACDA} vs {ENTFA, STAES, LISMO, GEOKA, BACSU, LACDA}, которая отсутствует в правильном дереве и отсутствует ветвь {LACDA, ENTFA} vs {CLOTE, BACSU, GEOKA, LISMO, STAES}, которая присутствует в правильном дереве. Кроме того, данное реконструированное дерево содержит ветвь {GEOKA, LISMO} vs {CLOTE, ENTFA, STAES, BACSU, LACDA}, которой нет в правильном дереве (в нём есть ветвь {GEOKA, BACSU} vs {CLOTE, ENTFA, STAES, LISMO, LACDA}).
Стоит отметить, что дерево, реконструированное с помощью алгоритма Neighbour Joining, является неукоренённым.
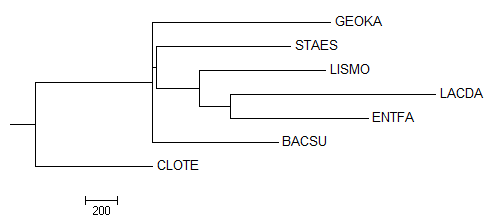
Average Distance Using BLOSUM62
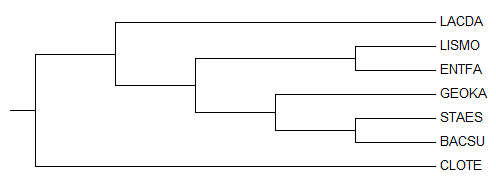
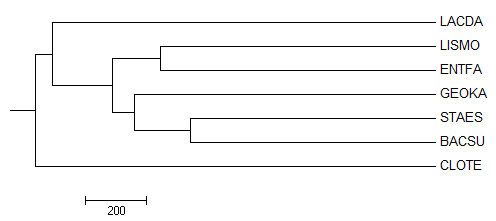
Все нетривиальные ветви этого дерева отличны от ветвей правильного дерева и перечислены ниже.
- {LACDA, CLOTE} vs {LISMO, ENTFA, GEOKA, STAES, BACSU}
- {LACDA, LISMO, ENTFA, CLOTE} vs {GEOKA, STAES, BACSU}
- {LACDA, LISMO, ENTFA, CLOTE, GEOKA} vs {STAES, BACSU}
- {LISMO, ENTFA} vs {LACDA, GEOKA, STAES, BACSU, CLOTE}
Neighbour Joining Using BLOSUM62
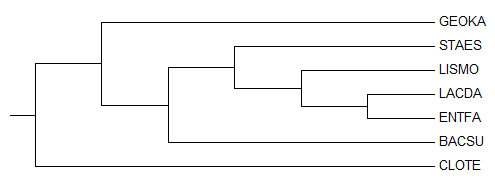
Из нетривиальных ветвей этого дерева (рассмотрено разрешение небинарного дерева, предложенное программой MEGA) лишь одна ветвь присутствует в правильном дереве: {LACDA, ENTFA} vs {GEOKA, STAES, LISMO, BACSU, CLOTE}. Три нетривиальные ветви, присутствующие в этом дереве и отсутствующие в правильном, приведены ниже.
- {GEOKA, CLOTE} vs {LISMO, ENTFA, GEOKA, STAES, BACSU}
- {GEOKA, CLOTE, BACSU} vs {STAES, LISMO, LACDA, ENTFA}
- {GEOKA, CLOTE, BACSU, STAES} vs {LISMO, LACDA, ENTFA}
Можно отметить, что реконструированное с помощью алгоритма Neighbour Joining дерево является неукоренённым.
Maximum Parsimony
Полученный .fasta-файл с выравниванием можно импортировать в программу MEGA, а затем реконструировать дерево методом Maximum Parsimony (в меню Phylogeny). Изображение дерева, укоренённого в одну из ветвей (Subtree > Root), приведено ниже.
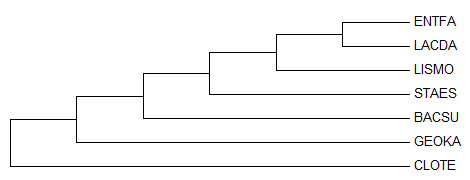
Это дерево имеет лишь одну нетривиальную ветвь, которая присутствует в правильном дереве: {LACDA, ENTFA} vs {GEOKA, STAES, LISMO, BACSU, CLOTE}. Три нетривиальные ветви этого дерева, которые отсутствуют в правильном дереве, приведены ниже.
- {ENTFA, LACDA, LISMO} vs {STAES, BACSU, GEOKA, CLOTE}
- {ENTFA, LACDA, LISMO, STAES} vs {BACSU, GEOKA, CLOTE}
- {ENTFA, LACDA, LISMO, STAES, BACSU} vs {GEOKA, CLOTE}
Сервис TreeTop
С помощью сервиса TreeTop можно реконструировать филогенетическое дерево по выравниванию. После ввода выравнивания в соответствующее поле после некоторого времени ожидания открывается страница с результатами, которые также можно выслать по адресу электронной почты. Страница с результатами содержит информацию о параметрах алгоритма, выравнивание, реконструированные деревья в текстовом и графическом форматах, а также матрицу расстояний и скобочные формулы реконструированных деревьев.
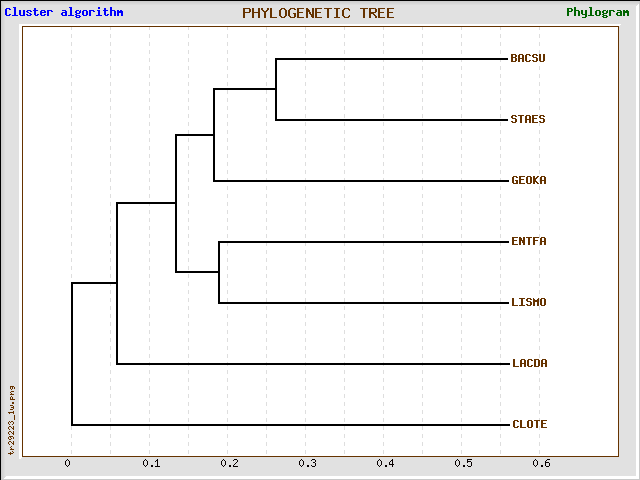
Реконструированное при помощи этого сервиса дерево (на изображении выше) для рассматриваемого выравнивания совпадает по топологии с деревом, реконструированным с помощью алгоритма Average Distance Using BLOSUM62 в JalView.
Ссылки
- Файл if2_bacteria.fasta.
- Файл if2_bacteria_alignment.fasta.
- Файл if2_bacteria_alignment.jar.
- Файл average_distance_using_percent_identity.tre.
- Файл neighbour_joining_using_percent_identity.tre.
- Файл average_distance_using_blosum62.tre.
- Файл neighbour_joining_using_blosum62.tre.
- Таксономический сервис NCBI.
- Сервис TreeTop, созданный в НИИФХБ имени А. Н. Белозерского.