
1. Нахождение гомологов белка в базе данных Swissprot
В этой части практикума осуществлялся поиск гомологов белка — с которым уже проводилась работа в 3 и 8 практикумах — при помощи Blast. Параметры, использующиеся при запуске, указаны в Таблице 1. Тексовая выдача доступна в файле align1.txt
| Параметр | Значение |
|---|---|
| Accession number(s), gi(s), or FASTA sequence(s) | P83597 |
| Job Title | P83597:RecName: Full=Antifungal peptide 2;... |
| Database | UniProtKB/Swiss-Prot(swissprot) |
| Organism | - |
| Exclude | - |
| Algorithm | blastp (protein-protein BLAST) |
| Max target sequences | 100 |
| Short queries | [selected] |
| Expect threshold | 10 |
| Matrix | BLOSUM62 |
| Gap Costs | Existance: 11 Extension: 1 |
| Compositional adjustments | Conditional compositional score matrix adjustment |
| Filter | Low complexity regions |
| Mask | - |
К сожалению (или к счастью), в выдаче оказалось только две последовательности, все от Эвкоммии вязолистной, при этом количество не менялось при изменении параметров запуска. Вероятно, этот ингибирующий рост грибов белок уникален для данного вида.
Также было выполнено выравнивание найденных последователбностей тем же способом, что описан в практикуме 10. Выравнивание можно найти в файле jalview_1.fasta, вывод программы Jalview представлен на Изображении 1 и в проекте jalview_1.jvp.
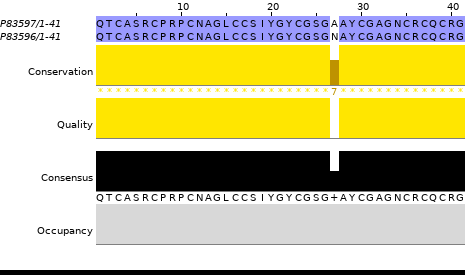
Как видно из выравнивания, последовательности определённо гомологичны, при этом крайне консервативны, о чём свидетельствует факт, что расхождение имеется только в замене одной аминокислоты. Ни инсерций, ни делеций не наблюдается.
2. Поиск в Swissprot гомологов последовательности зрелого белка MERS-CoV
В 9 практикуме было проведено выделение из последовательности полипротеина (Replicase polyprotein 1ab) в файл str.fasta последовательности ингибитора трансляции nsp1 (Host translation inhibitor nsp1, координаты: 1-193).
Здесь уже описанным выше способом проводится поиск гомологов и последующее выравнивание 5 находок (AC: K9N638, P0C6T5, P0C6F7, P0C6W4, P0C6W1) из 54 (все их можно найти в файле align3.txt, в списке их 55, но искомую не учитываем). В выдаче Jalview были удалены все буквы до первой (не потребовалось) и последней выровненных с другими букв. Результат представлен на Изображении 2 и в проекте align11.jvp.
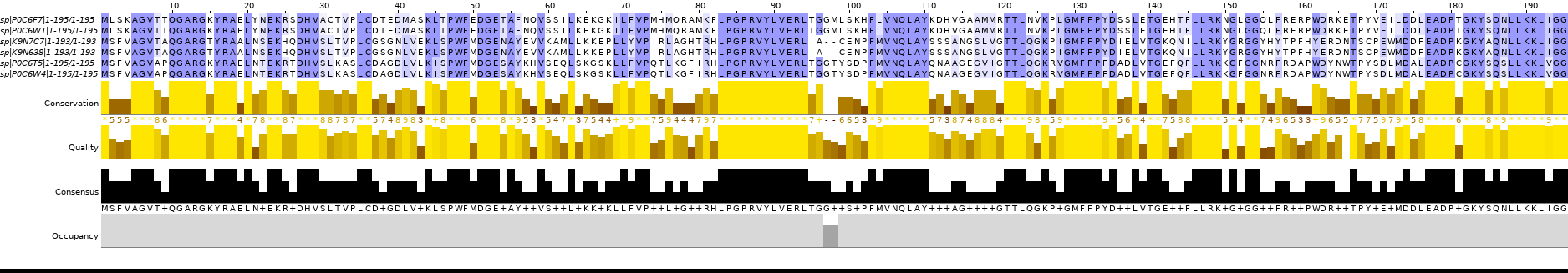
Судя по количеству и длине консервативных участков, последовательности можно считать гомологичными и достаточно консервативными.Тот же вывод позволяет сделать значение E-value от 9e-133 до 6e-59. Вероятно, либо последовательности взяты из родственных вирусов, либо белок играет очень важную функцию, что не позволяет допускать сильные вариации.
3. Исследование зависимости E-value от объёма банка
Был проведёт второй сеанс поиска с указанием организма (MERS-CoV (taxid:1335626)). При этом количество находок сократилось до двух (K9N638, K9N7C7), а их E-value уменьшилось с 9e-133 и 2e-132 до 7e-137 и 2e-136 соответственно.
Результаты подтверждают, что при уменьшении размера базы данных поиска E-value также уменьшается. Эта зависимость обусловлена теоремой Карлина: E-value = Kmn·e-λS, где S – вес, m – длина исходной последовательности, n – размер базы данных (суммарная длина всех последовательностей), K и λ – константы. Это уравнение может помочь вычислить примерную долю белков MERS-CoV через изменение E-value:
E-value1=mn1*2-B
E-value2=mn2*2-B
n1/n2=E-value1/E-value2
По среднему значению из изменений двух находок в Swissprot примерно 0.009% – белки MERS-CoV.