Системы рестрикции-модификации (Р-М) - это один из механизмов защиты прокариот от чужеродной ДНК, например, бактериофагов.
Система Р-М умеет распознавать определенные короткие последовательности ДНК (сайты рестрикции) и гидролизовать ДНК, если эти последовательности не метилированы.
В ДНК клетки все сайты рестрикции заметилированы, а в ДНК фагов - нет. Поэтому клеточная ДНК остается невредимой, а ДНК фагов гидролизуется.
Но иногда в процессе метилирования сайтов рестрикции случаются ошибки и геном бактерии может быть гидролизован.
Из-за этого бактерии выгодно содержать в геноме как можно меньше сайтов узнавания систем Р-М, чтобы уменьшить вероятность случайного гидролиза.
Отбор против сайтов в геноме можно обнаружить, сравнив наблюдаемое число сайтов с ожидаемым числом.
Например, можно вычислить отношение наблюдаемое/ожидаемое число сайтов (контраст), и если это отношение меньше 1, значит встретилось меньше сайтов, чем ожидалось.
Обнаружив такие "избегаемые" сайты можно предсказать, какие системы Р-М бактерия содержит (или содержала в недавнем прошлом).
Бактерии достаточно быстро меняют набор систем Р-М. Поэтому даже бактерии (или археи) одного вида могут содержать разный набор систем Р-М в разных популяциях.
Наша задача - сравнить предполагаемые (по избеганию сайтов) наборы систем Р-М в полном геноме бактерии из NCBI и наборе контигов того же вида из метагенома кишечника человека.
Задание 1
Для работы я выбрала бактерию, обитающую и воспользовалась списком всех известных сайтов рестрикции-модификации второго типа.
С помощью веб-сервиса, рекомендованного в практикуме, я исследовала сайты рестрикции-модификации, представленные в данной бактерии:
выходной файл
Следующий этап этого задания - проделать аналогичную работу с набором контигов той же бактерии из метагенома кишечника человека.
выходной файл
Стоит отметить, что в обоих случаях было найдено 2 сайта рестрикции, против которых производится отбор
Для удобства сравнения сайтов рестрикции я свела их в одну таблицу
У этих бактерий был найден один общий сайт и один различающийся сайт.
Задание 2
Последовательность Шайн — Дальгарно — это рибосомальный сайт связывания на прокариотической мРНК, обычно на расстоянии около 10 нуклеотидов до старт-кодона AUG.
В данном задании я производила поиск такой последовательности
Сначала я скачала fasta-файл с полной последовательностью бактериальной хромосомы и файл, содержащий информацию о CDS на этой хромосоме.
Для дальнейшего анализа мною было выбрано 200 самых длинных кодирующих последовательностей.
Итак, для этих последовательностей нужно найти участки, в которых имеет смысл искать последовательность Шайн - Дальгарно.
Далее я получила последовательности с указанными координатами.
Теперь появилась возможность искать в этих нуклеотидных фрагментах последовательность Шайн-Дальгарно
Последовательности Шайн-Дальгарно искались сначала на 200 последовательностях с помощью MEME
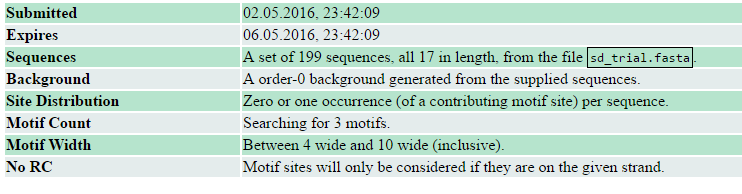
Таблица 1 Параметры запуска MEME
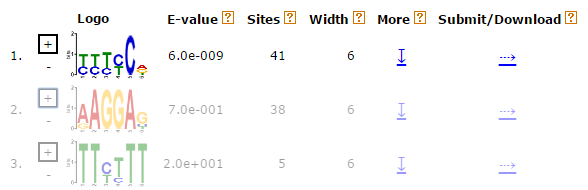
Рисунок 1 Мотивы
Как видно из рисунка, мотив, похожий на Шайн-Дальгарно, был вторым по "популярности". Однако, его e-value незначительно ниже первого. Найденную PWM я использовала для поиска этого мотива уже по всем генам бактерии, предварительно расширив область поиска до 21 нуклеотида. Порог p-value составлял 0.001, поиск производился только на данной цепи

Таблица 2 FIMO
Сходный мотив нашёлся в 1906 последовательностях из 2470.
выходной файл
Задание 3
Был проведен контроль качества ридов с помощью команды fastqc chipseq_chunk27.fastq:
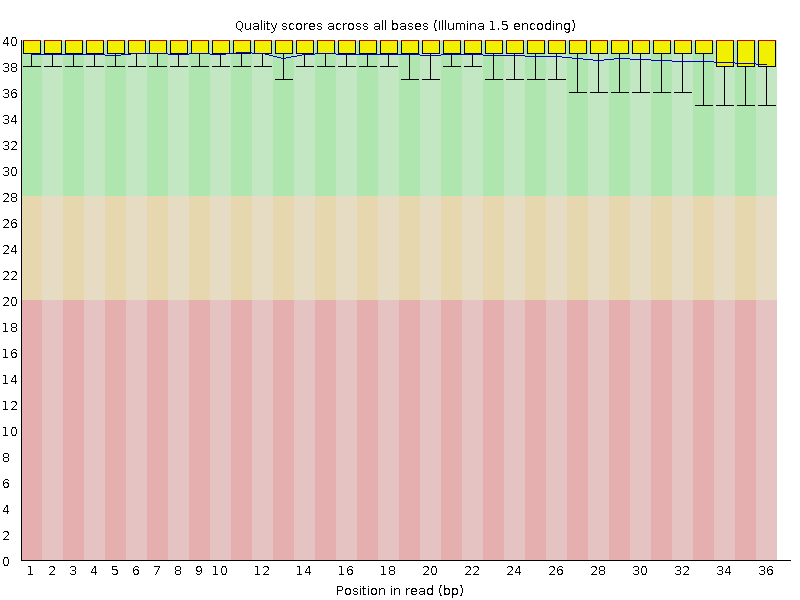
Рисунок 2 Контроль качества чтений
Всего ридов 7832. Качество чтений очень хорошее, поэтому программа trimmomatic для очистки от чтений с плохим качеством не применялась.
Картирование прочтений на геном человека hg19 выполнялось с помощью программы BWA
Для последующего анализа полученого выравнивания я воспользовалась программой samtools и предприняла следующие действия:
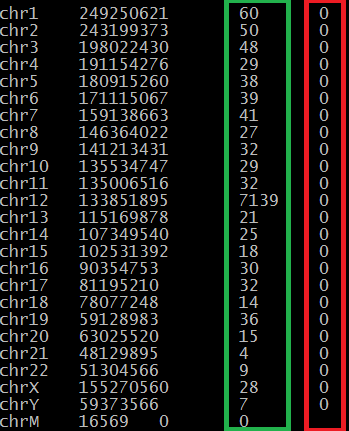
Рисунок 3 Результат
Следующий этап работы - поиск пиков (peak calling). В нашем случае пики - это небольшие участки ДНК, на которые картировалось много ридов, что говорит о том, что белок в ChIP-seq связывался именно c этими участками.
Для поиска пиков я воспользовалась программой MACS:
macs2 callpeak -t out.predix.bam
С настройками по умолчанию эта команда не нашла пиков. Поэтому она была запущена со следующими параметрами:
macs2 callpeak -n chunk27 -t out.predix.bam --nomodel
Файл .narrowPeak был визуализирован с помощью UCSC Genome Browser. Для этого в начало файла были добавлены строки:
track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="Peaks from chunk 27" browser position chr12:56224436-56862474.
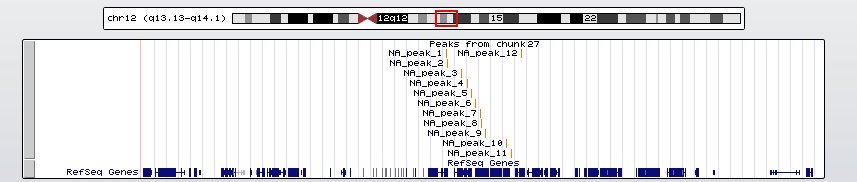
Рисунок 4 Пики
Пик 9
Пик 12
Задание 4
TATA-бокс связывающий фактор TBP - архейный и эукариотический белок, узнающий восьминуклеотидный сигнал в ДНК с консенсусом TATAWAAR.
Он является одним из ключевых ДНК-узнающих белков при образовании на промоторе генов комплекса TFIID инициации транскрипции с помощью Pol II.
Тем не менее, лишь часть промоторов имеет сигнал TATA-box, связываемый TBP. Цель этого задания - найти в геноме человека три гена, имеющих такой сигнал, и один ген, не имеющий сигнала.
Я производила поиск при помощи сервиса UCSC GenomeBrowser. Там я нашла белок TBP (TATA binding protein) и выбрала эксперимент ChiP-Seq, проводившийся на клеточной линии GM12878.
В эксперименте использовались антитела к TBP ab62126.
Первый ген - ген ГМФ-связывающей фосфодиэстеразы. Положение в геноме - 120415550-120549981, 4 хромосома, + - цепь. Длина - 7005 нуклеотидов.
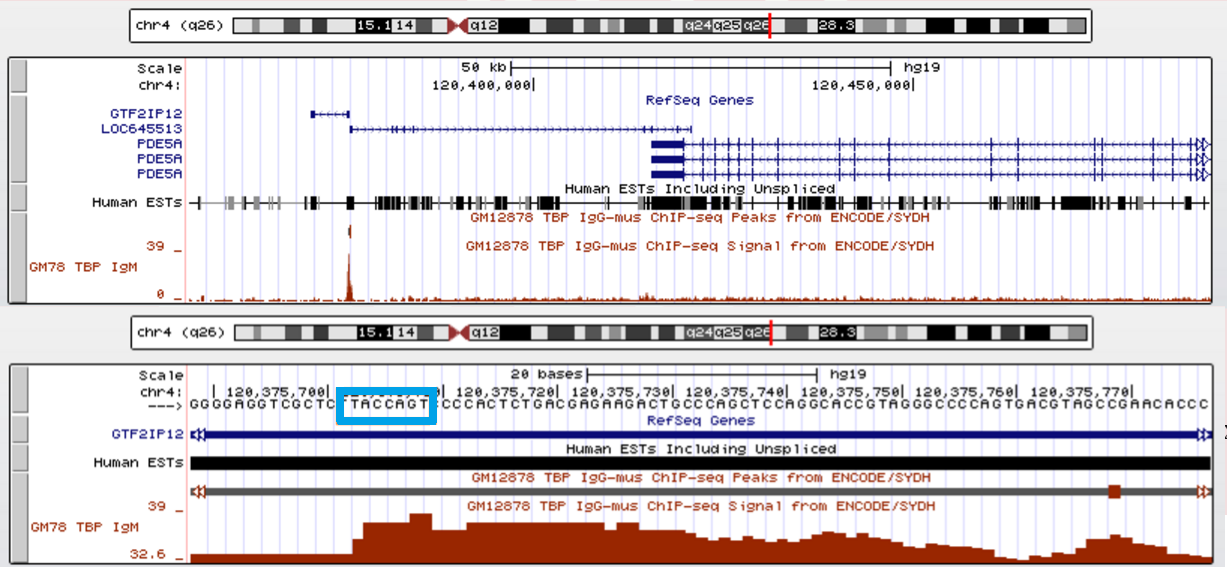
Рисунок 5 Ген PDE5A Сверху - в мелком масштабе Снизу - в крупном масштабе. Последовательность выделена голубой рамкой
Второй - ген одной из каталитических субъединиц белка, катализирующего гидролиз глюкозо-6-фосфата. Белок локализован в ЭПР. Положение в геноме - 42148098-42153712 на 17 хромосоме (+ - цепь). Длина последовательности - 6515 нуклеотидов.
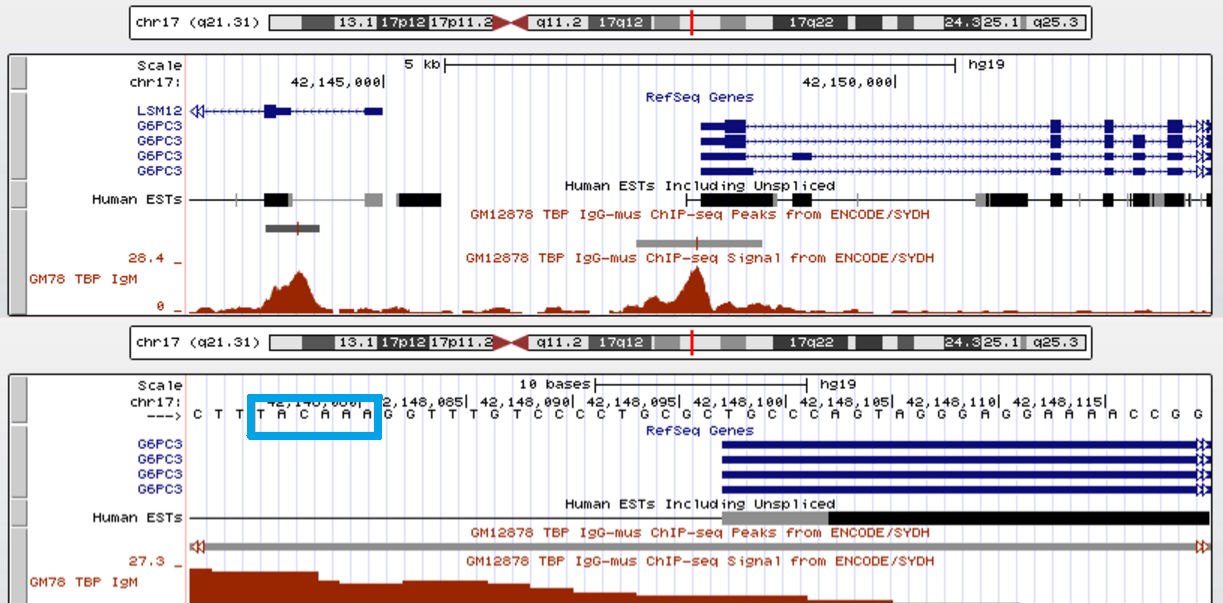
Рисунок 6 Ген G6РС3 Сверху - в мелком масштабе Снизу - в крупном масштабе. Последовательность выделена голубой рамкой
Третий - ген человеческого белка гликогенина - 1. Это гликозилтрансфераза, которая катализирует формирование небольших глюкозных полимеров. Размер кодирующей последовательности составляет 35351 нуклеотидов. Позиция в геноме - 148709428-148744778, на 3 хромосоме (+ - цепь). Имеет 6 экзонов.
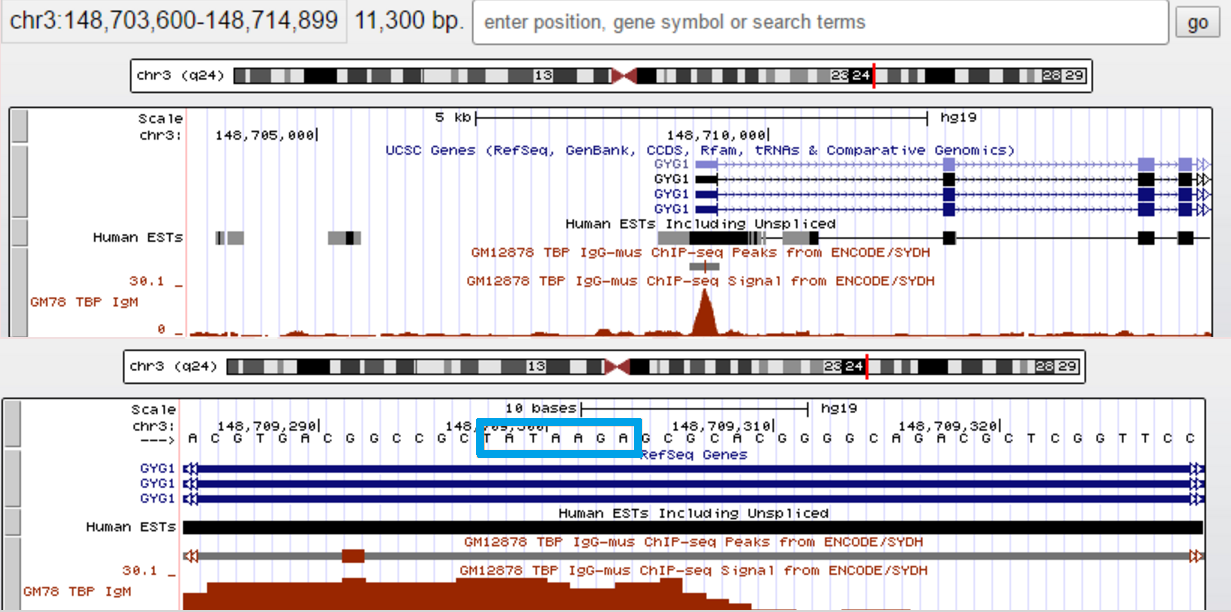
Рисунок 7 Ген GYG1 Сверху - в мелком масштабе Снизу - в крупном масштабе. Последовательность выделена голубой рамкой
Четвёртый - Этот белок - рецептор ангиотензина-2 первого типа. Находится на + - цепи третьей хромосомы. Положение на хромосоме - 148415658-148460790. Длина гена - 2272 нуклеотида.
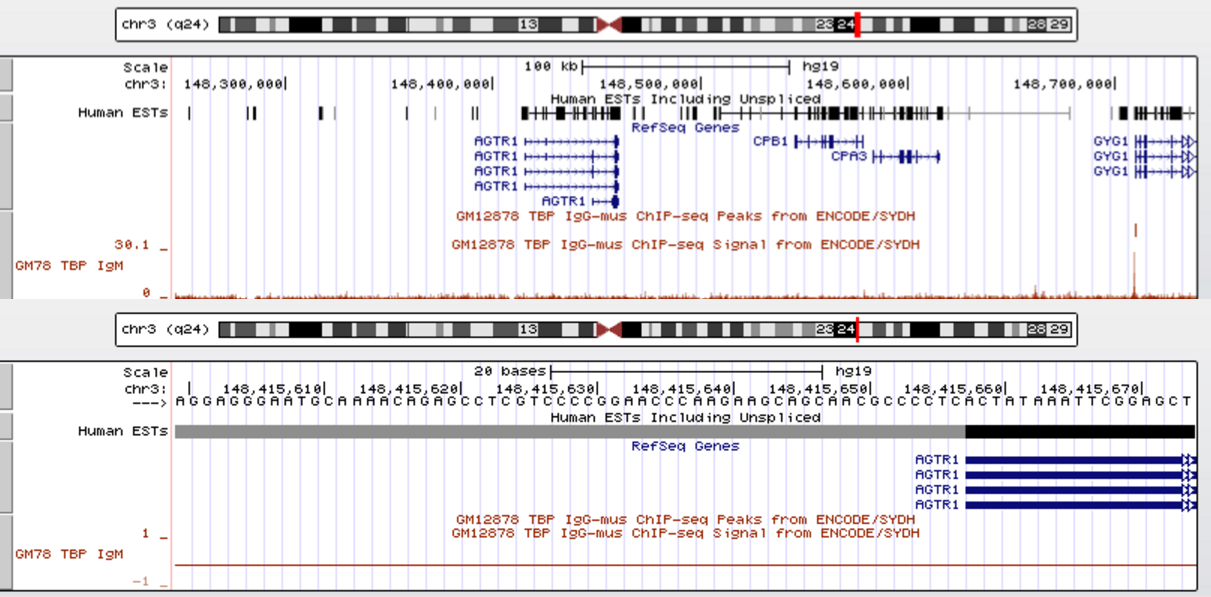
Рисунок 6 Ген AGTR1 Сверху - в мелком масштабе Снизу - в крпном масштабе.