Поиск сигналов. chip-seq
Дано: Файл chipseq_chunk6.fastq с ридами Illumina, полученные в результате ChIP-seq эксперимента
С помощью программы FastQC был выполнен контроль качества чтений. FastQC принимает на фход файл с ридами,
проволит анализ качества и приводит обширный отчет, состоящий из нескольких модулей (fastqc.html).
График "Per base quality" приведен на рис.1.
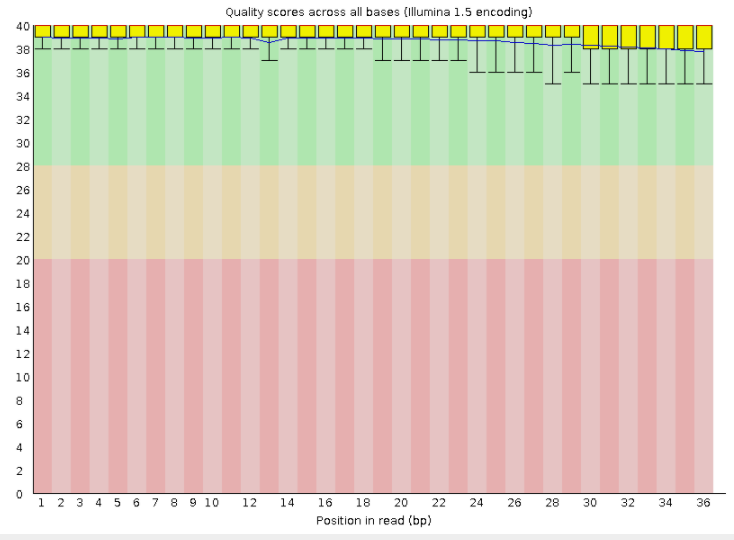 |
| Рис. 1. Качетво чтений (FastQC) |
Всего в файле 7832 ридов, длина которых составляет 36 п.н. Исходные риды имеют хорошее качество - все позиции лежат в зеленой области. Поэтому очистка чтений не проводилась.
После этого было проведено картирование прочтений на геном селовека hg19 с помощью программы BWA:
bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk6.fastq > chipseq_chunk6.sam
Анализ полученных результатов проводился с помощью пакета samtools:
Перевод выравнивания в бинарный формат:
samtools view -b -o chipseq_chunk6.bam chipseq_chunk6.sam
Сортировка выравнивания по координате начала чтения в референсной последовательности:
samtools sort chipseq_chunk6.bam -T chip_temp -o chipseq_chunk6.sorted.bam
Индексирование отсортированного файла:
samtools index chipseq_chunk6.sorted.bam
Подсчет чтений, откартировавшихся на геном (результат: chipseq_chunk6.idxstats):
samtools idxstats chipseq_chunk6.sorted.bam > chipseq_chunk6.idxstats
Общее количество ридов - 7832. Все они откартировались на геном, причем большая часть - на chr12. Поэтому, скорее всего прочтения именно с этой хромосомы
были предложены для анализа.
Поиск пиков осуществлялся с помощью программы MACS(Model-based Analysis of ChIP-Seq):
macs2 callpeak -t chipseq_chunkX.sorted.bam --nomodel
В итоге работы программы было получено три файла, содержащих информацию о найденных пиках : NA_peaks.narrowPeak,
NA_peaks.xls, NA_summits.bed. Часть информации из файла NA_peaks.xls представлена в табл. 1.
Всего было найдено 12 пиков. Наиболее достоверными являются пики №8 и №9 (т.к. значения log10(pvalue) и log10(qvalue) - наибольшие). эти же пики являются самыми
широкими (их ширина составляет 340 и 344 п.н., соответственно). Наименее достоверные пики - №7 и №10.
Таблица 1. Пики, найденные с помощью программы MACS
| chr |
start |
end |
length |
abs_summit |
pileup |
-log10(pvalue) |
fold_enrichment |
-log10(qvalue) |
name |
| chr12 |
56224436 |
56224703 |
268 |
56224592 |
22.00 |
15.78747 |
7.37180 |
9.40817 |
NA_peak_1 |
| chr12 |
56236669 |
56236942 |
274 |
56236779 |
39.00 |
32.92253 |
11.36364 |
26.10617 |
NA_peak_2 |
| chr12 |
56349925 |
56350140 |
216 |
56350032 |
23.00 |
21.44869 |
10.34483 |
14.89172 |
NA_peak_3 |
| chr12 |
56404010 |
56404274 |
265 |
56404130 |
19.00 |
14.47894 |
7.40741 |
8.18992 |
NA_peak_4 |
| chr12 |
56440893 |
56441142 |
250 |
56441017 |
29.00 |
26.81850 |
11.45038 |
20.13817 |
NA_peak_5 |
| chr12 |
56473687 |
56473986 |
300 |
56473848 |
33.00 |
28.93972 |
11.25828 |
22.21293 |
NA_peak_6 |
| chr12 |
56510250 |
56510479 |
230 |
56510302 |
25.00 |
16.07615 |
6.80628 |
9.69051 |
NA_peak_7 |
| chr12 |
56521520 |
56521859 |
340 |
56521702 |
93.00 |
83.42138 |
15.98639 |
76.07829 |
NA_peak_8 |
| chr12 |
56551934 |
56552277 |
344 |
56552107 |
125.00 |
131.48038 |
23.07692 |
122.00696 |
NA_peak_9 |
| chr12 |
56730446 |
56730665 |
220 |
56730545 |
29.00 |
16.15354 |
6.04839 |
9.76505 |
NA_peak_10 |
| chr12 |
56771137 |
56771361 |
225 |
56771215 |
20.00 |
16.40781 |
8.26772 |
10.00295 |
NA_peak_11 |
| chr12 |
56862248 |
56862474 |
227 |
56862349 |
20.00 |
15.77306 |
7.89474 |
9.39724 |
NA_peak_12 |
Полученная информация о пиках была визуализирована с помощью сервиса UCSC Genome Browser.
В качестве входного файла использовался файл NA_peaks.narrowPeak, но в начало были добавлены слудеющие строки:
track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="Peaks from chunk 6"
browser position chr12:56224435-56862474
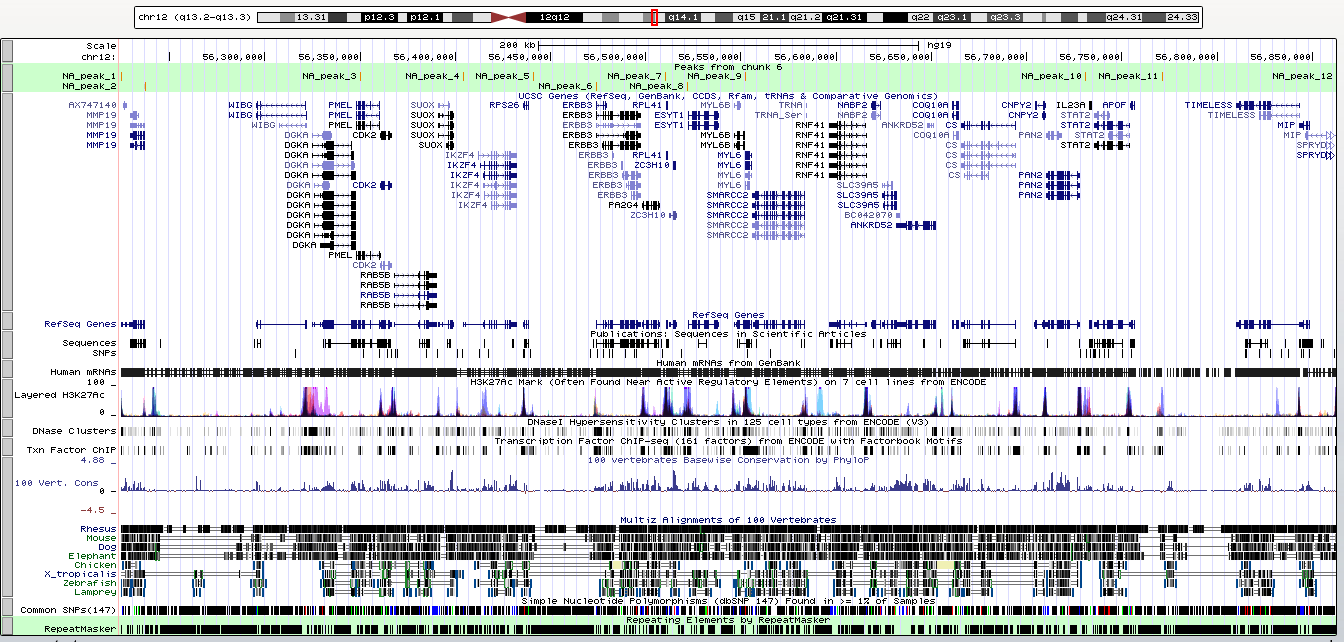 |
| Рис. 2.Визуализация с помощью UCSC Genome Browser |