Anna Zheltova
Seventh term (Седьмой семестр):
Electron density in PyMol (Электронная плотность в PyMol)
Crystal recovery in PyMol (Восстановление кристалла в PyMol)
Expansion of a function in a Fourier series (Разложение функции в ряд Фурье)
Analysis of the NMR file (Анализ ЯМР-файла)
Definition of the secondary structure (Определение вторичной структуры)
Locating hydrophobic clusters (Нахождение гидрофобных кластеров)
Surface construction and analysis in PyMOL (Построение и анализ поверхностей в PyMOL)
Совмещение структур
Поиск структурных гомологов белка 1DUP
Использовали поиск по сходству структур в PDBeFold.
Были найдены структурные гомологи для цепи А (для всего белка, так как белок содержит 1 цепь) с RMSD между 0,8 и 2,5 и длиной выравнивания более 50% от длины 1DUP. Выбранные гомологи представлены в таблице.
Таблица 1. Выбранные гомологи.

Совмещение структур белка и его структурных гомологов
Далее с помощью PDBeFold было получено множественное выравнивание.
Результаты структурного выравнивания (выравнивания последовательноcтей) предствалены в данном файле.
Результаты структурного выравнивания (файл с совмещёнными сруктурами) предствалены в данном файле.
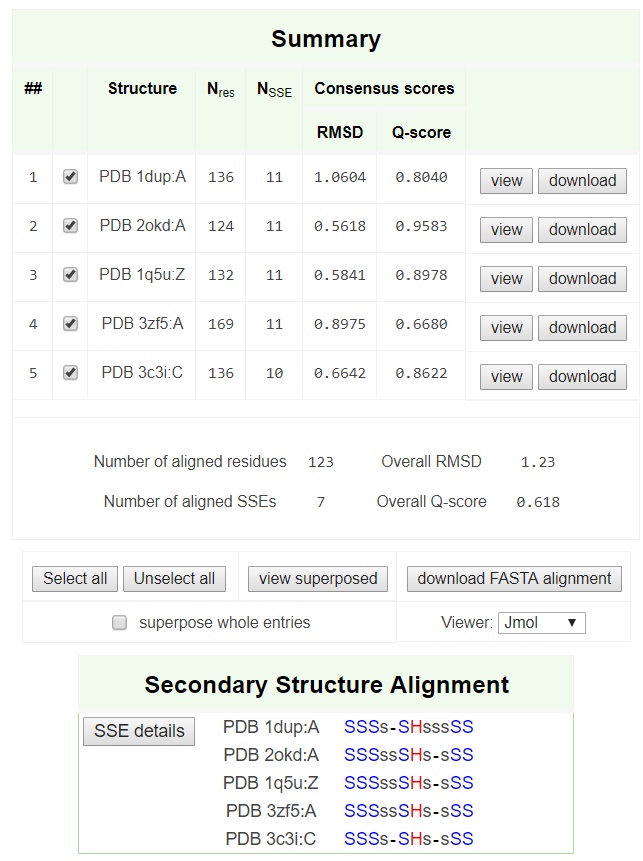
Таким образом были совмещены 5 структур. Результат совмещения представлен на рисунке:
Для дальнейшей работты в PyMol был установлен Plugin CONSCRIPT, файл с плагином .
Для получения изображения совмещения и дальнейшей работе с ним был открыт файл в PyMol с совмещенными структурами и выполнены следующие команды:
extract 1dup, chain A
extract 3zf5, chain D
extract 3c3i, chain E
extract 1q5u, chain C
extract 2okd, chain B
show cartoon
hide all
show cartoon
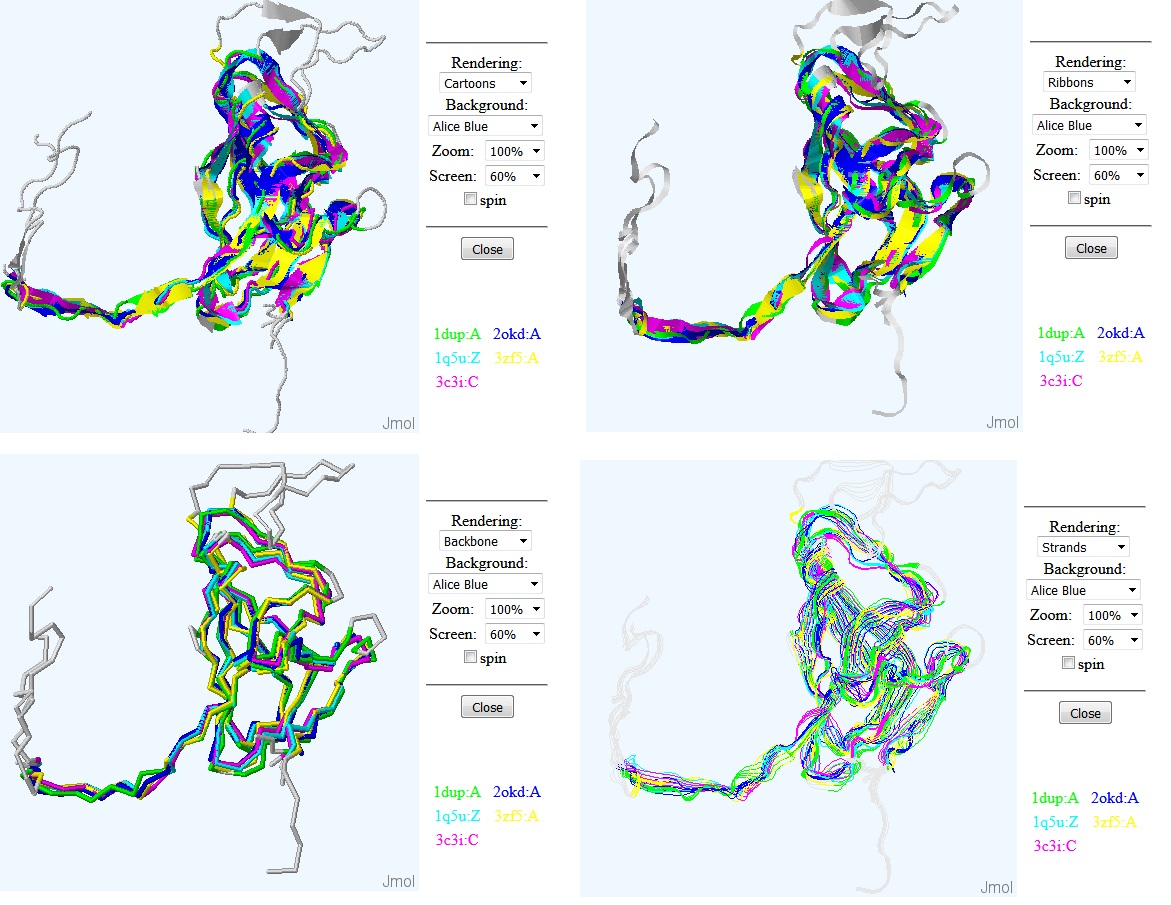
В принципе структуры совмещаются хорошо, общую картину портит 3zf5.
Были найдены отличия выравнивания последовательностей по структуре от выравнивания тех же последовательностей, построенного программой множественного выравнивания MAFFT.
Выравнивание PDBeFold по структурному совмещению, визуализированое с помощью Jalview (изображение в полном размере доступно по этой ссылке):
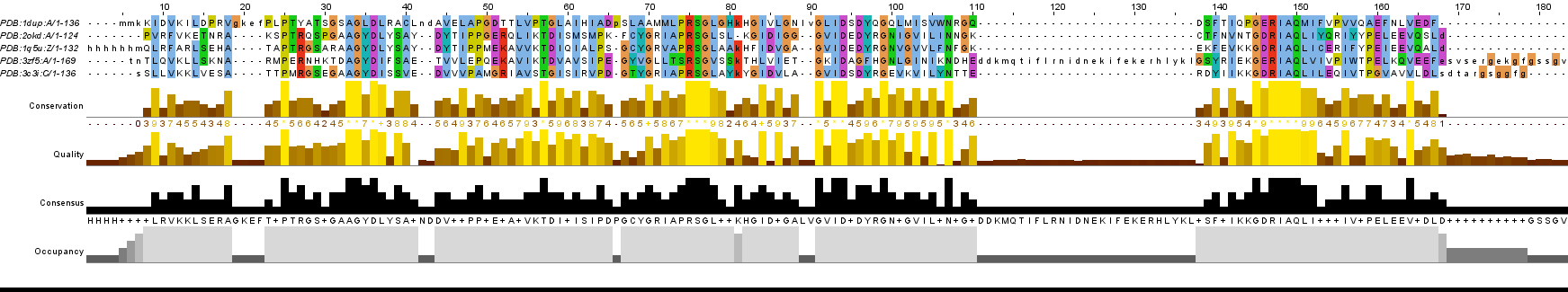
Выравнивание последовательностей с помощью MAFFT, визуализированое в Jalview (изображение в полном размере доступно по этой ссылке):

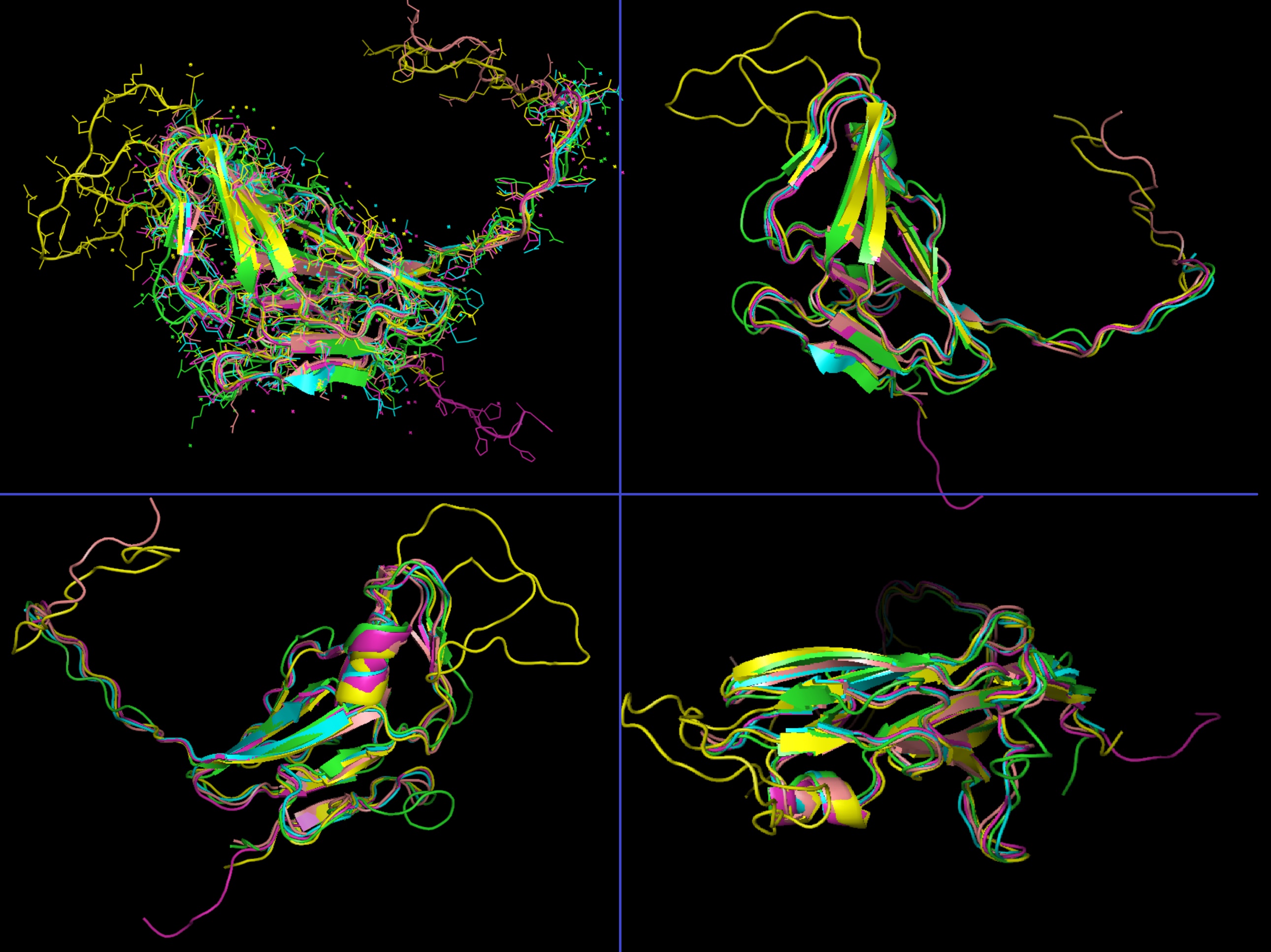
Отличия в выравниваниях выделены на рисунке:
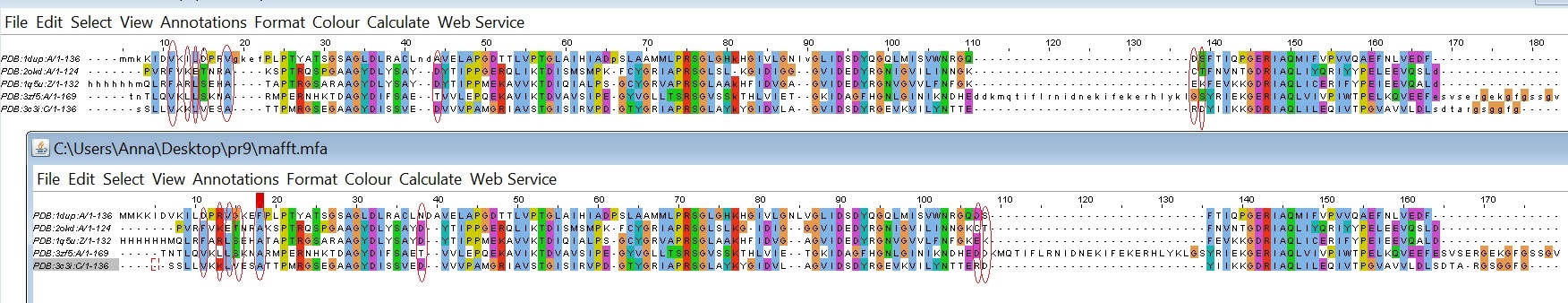
В выравниваниях, построенных разными методами наблюдается немного отличий. Основное отличие связано с началом выравнивания (выделено на предыдущем рисунке) и несколько отличий обнаружено в средине выравнивания (также выделены на предыдущем рисунке).
Для того, чтобы разобраться, кто прав, перейдем к визуализации:

Оставшиеся различия никак не влияют на вывоД: выравнивание с помощью PDBeFold более достоверное. Оно намного лучше соответствует положению аминокислотных остатков в белке. Программа множественного выравнивания MAFFT в некоторых случаях приводит к ошибочному выравниванию, так как расположенные рядом остатки могут оказаться в разных колонках..
Совмещение по заданному выравниванию
Была выбрана одна структура (1BD2, region d:118-203) константного домена T-клеточного рецептора из цепи альфа и одна (1QSE, region e:119-246) из цепи бета.
Были построены карты бета-листов при помощи SheeP.
Карта бета-листов альфа цепочки 1BD2:
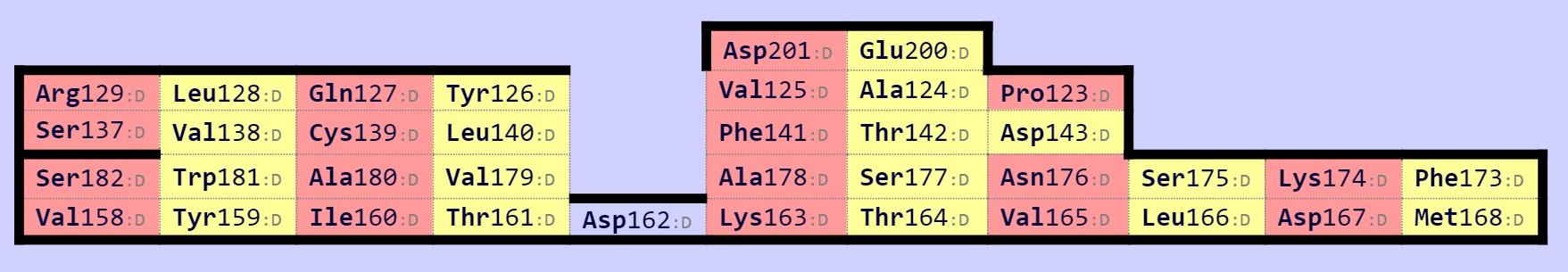
Карта бета-листов бета цепочки 1QSE:

В каждой карте был найден консервативный остаток цистеина, образующий дисульфидную связь. В альфа-цепи 1BD2 - Cys 139, в бета-цепи 1QSE - Cys 147
Было построено выравнивание бета-листов. Консервативные цистеины выровненны, и их выравнивание задает выравнивание всего центрального тяжа. Остатки, спаренные с консервативным цистеином, также считаются выровненными, они задают выравнивание еще каких-то тяжей. Так было построено выравнивание целых листов.
Команда для PyMol, которая совмещает структуры по данному выравниванию:
fetch 1bd2_chain_D
fetch 1qse_chain_E
hide all
select first, 1bd2_chain_D and resi 126+128+138-140+179-181 and name CA
select second, 1qse_chain_E and resi 128+130+146-148+193-195 and name CA
pair_fit first, second
При выравнивании RMS = 0.566
Полученно совмещение представлено на рисунке (красный цвет - альфа-цепь 1BD2, зеленый - бета-цепь 1QSE):
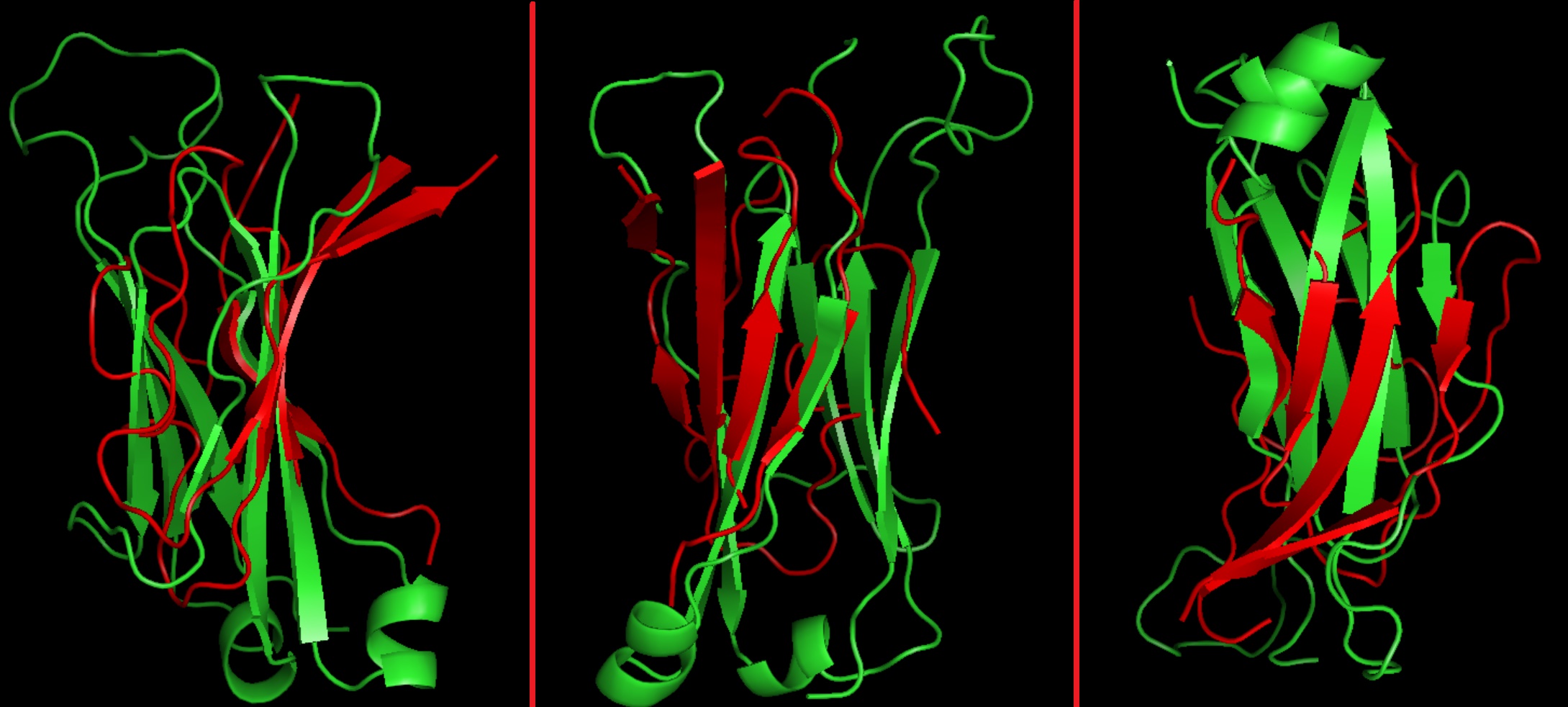
Бета-тяжи имеют одинаковое направление, ход полипептидной цепи в пространстве аналогичен для бета-тяжей, но при этом абсолютно различен для петель (топология петель не совпадает). Однако несмотря на то, что некоторые бета-тяжи совместились нормально, выравнивание не является хорошим, на мой взгляд.
Пара белков, для которых гибкое структурное выравнивание включает существенно больше остатков, чем жесткое
Был взят пример из статьи Flexible structural protein alignment by a sequence of local transformations, doi:10.1093/bioinformatics/btp296.
Сравнение рибосомного белка L1 мутанта S179C (PDB:1AD2) и 50S рибосомального белка L1P Methanococcus jannaschii (PDB:1CJS).
Для визуализации примера были использованы PDBeFold и FATCAT .
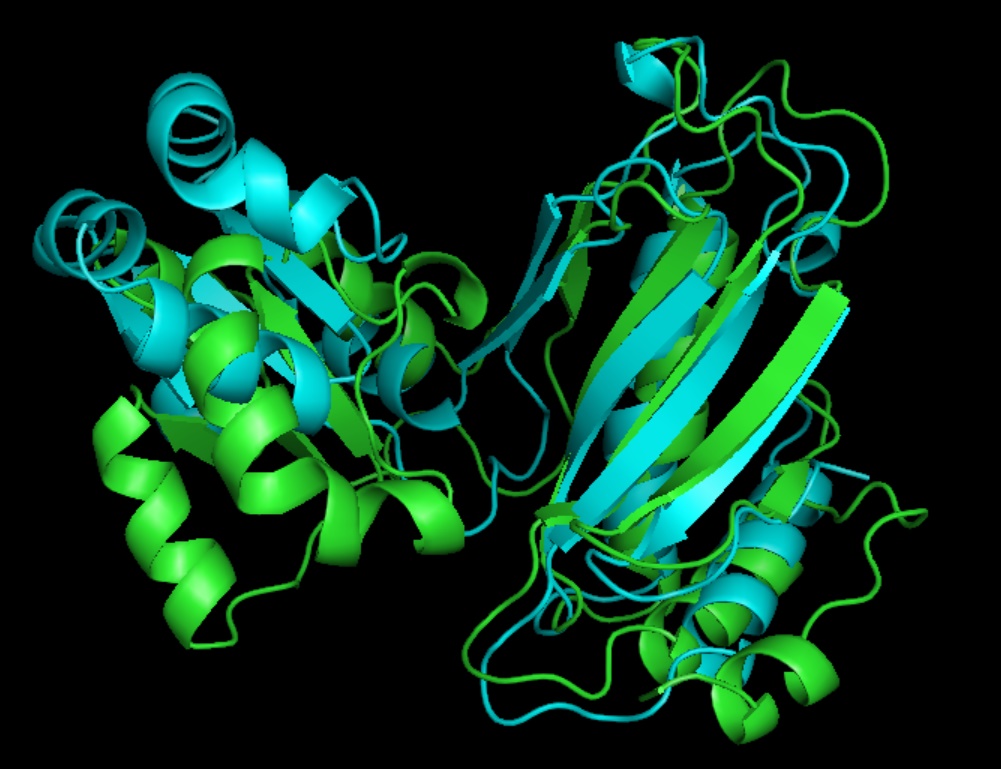
Жесткое выравнивание
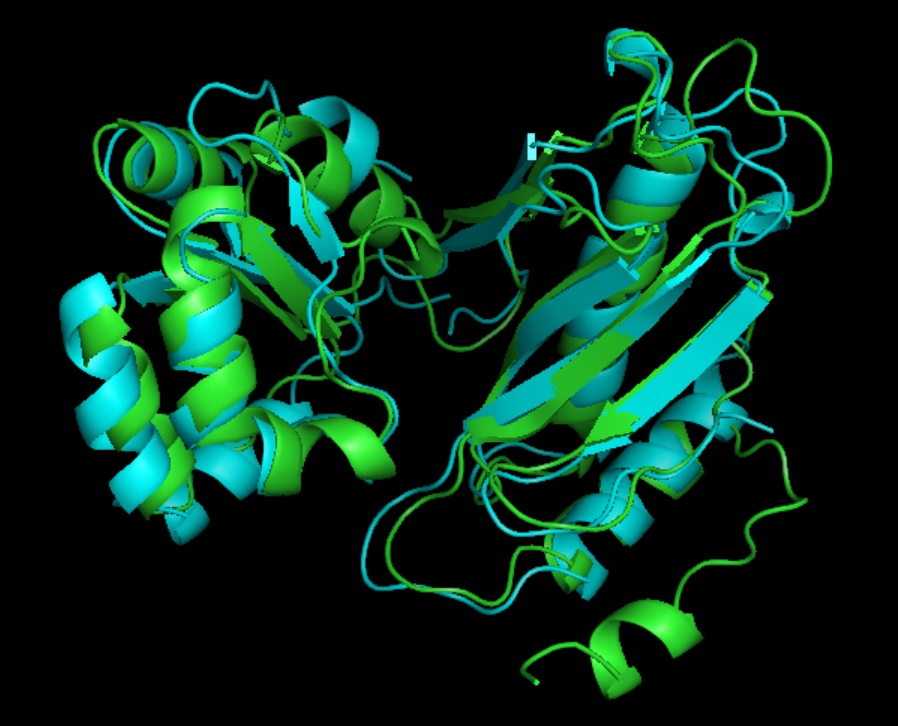
FATCAT. Гибкое выравнивание.
Для жесткого выравнивания только один домен из каждого белка хорошо выравнивается. Однако несколько локальных преобразований (представлены на рисунке ниже) позволяют значительно улучшить суперпозицию
