|
|
|
|
|
|
Блок 1: структуры нуклеиновых кислот. Комплексы ДНК-белок.
Задание 1.
Краткое описание структуры ДНК
Для исследования были выбраны цепи A и B белка и цепи C и D, представляющие ДНК со следующей последовательностью:
цепь C [1001] 5' - TCAGTCTAGACATAC - 3' [1015]
цепь D [2015] 3' - AGTCAGATCTGTATG - 5' [2001]
Функции белка: Модулятор траснкрипции, активируемый ТФР-бета (трансформируемым фактором роста) и активин-рецептороподобной киназой 1. Рецепторорегулируемый SMAD.
Исследование структуры ДНК C помощью программ find_pair и analyze были определены средние значения торсионных углов. По этим значениям данная ДНК очень похожа на В-форму, хотя наблюдаются отклонения у углов гамма и бета. Ссылка на Excel-файл.
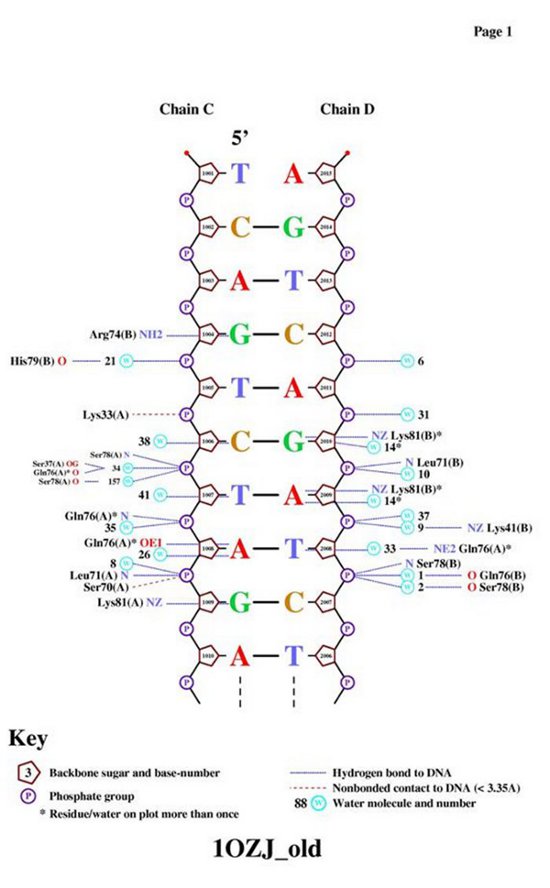
Исследование природы ДНК-белковых контактов
Скрипт, выделяющий заданные группы атомов. Изменяя параметры оператора within можно выделять атомы, между которыми возможны интересующие нас контакты. Здесь выделены неполярные контакты между атомами азотистых оснований, обращенных в сторону малой бороздки и атомами ДНК.
| Контакты атомов белка: | Полярные | Неполярные | Всего |
| с остатками 2'-дезоксирибозы | нет | 9 | 9 |
| с остатками фосфорной кислоты | 9 | 10 | 19 |
| с остатками азотистых оснований со стороны большой бороздки | 7 | 5 | 12 |
| с остатками азотистых оснований со стороны малой бороздки | 2 | нет | 2 |
Резюме:
1) Больше всего контактов с белком образуют остатки фосфорной кислоты.
2) Меньше всего контактов в белком образуют атомы азотистых оснований со стороны малой бороздки. Со стороны большой борозки контактов больше - это может быть обусловлено пространственным расположением атомов: больше свободного пространства для возможноых контактов именно со стороны большой бороздки.
Получение схемы ДНК-белковых контактов с помощью nucplot
Характеристика ДНК-связывающего домена MH1
SMAD3 состоит из двух доменов - MH1 и MH2.
Домен MH1 (MAD homology 1) расположен на N-конце белков, родственных MAD, таких как Smadы.
Этот домен отделён от домена MH2 неконсервативным связывающим участком. Из кристаллической структуры MH1 видно, что высоко консервативная бета-"шпилька" из 11 остатков связывает оптимальную последовательность ДНК GNCN (богатую гуанином и цитозином) в районе большой бороздки, что необходимо для активации транскрипции целевых генов. Не все MH1, однако, могут связываться с ДНК. SMAD2 неспособен связывать ДНК и содержит в "шпильке" крупный участок, препятствующий этому связыванию. Основная спираль (H2) в MH1 с сигналом локализации в ядре KKLKK, как выявлено, необходима для импорта Smad3 в ядро. Smadы также используют MH1-домен для взаимодействия с факторами транскрипции, такими как Jun, TFE3, Sp1 и Runx.
Задание 2.
Для исследования была выбрана цепь B, представляющая тРНК со следующей последовательностью:
[501]
5' - CCGGCGGUAGUUCAGCCUGGUAGAACGGCGGACUGUAGAUCCGCAUGUCGCUGGUUCAAAUCCGGCCCGCCGGACCA - 3' [577]
На 3' -конце присутствует триплет CCA, но координаты его атомов не приведены.
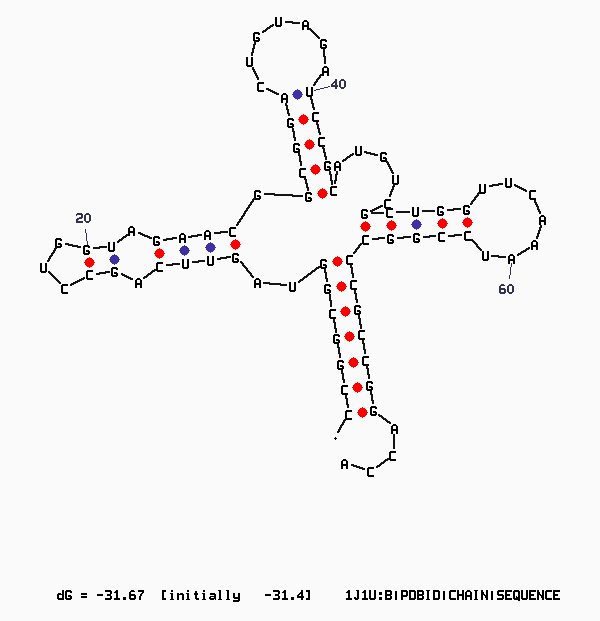
Исследование вторичной структуры.
С помощью программы find_pair пакета 3DNA были определены возможные водородные связи между азотистыми основаниями.
акцепторный стебель выделен красным, Т-стебель - зеленым, D-стебель - синим, антикодоновый - оранжевым (антикодон GUA(535-537) - жёлтый)
Вторичная структура тРНК из METHANOCALDOCOCCUS JANNASCHII
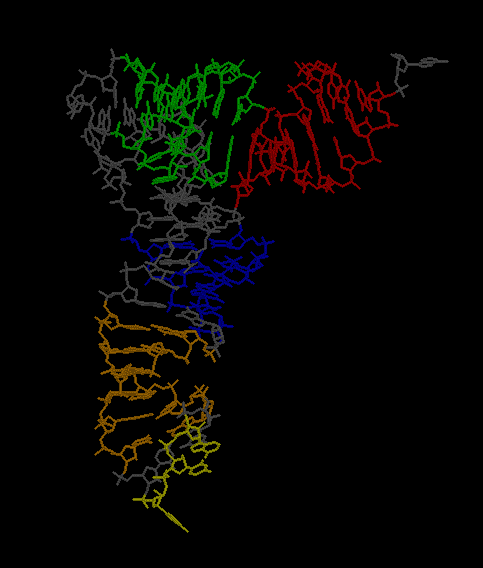
Скрипт для получения изображения: тут
Структуру стеблевых дуплексов поддерживают 21 каноническая и 3 неканонические пары оснований. (G564-U552, G527-A545 и A539-C533)
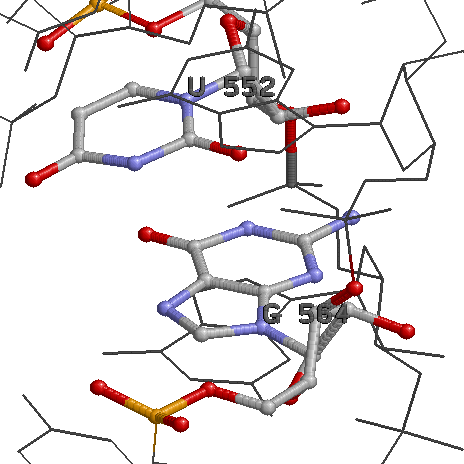
Не обнаружено ни остатков тимидина, ни дигидроуридинов. Вариабельная петля не выражена.
Исследование третичной структуры.
1.Стекинг-взаимодействия между основаниями
Т-стебель - акцепторный стебель (перекрывание 3.74) 550-566, 507-567
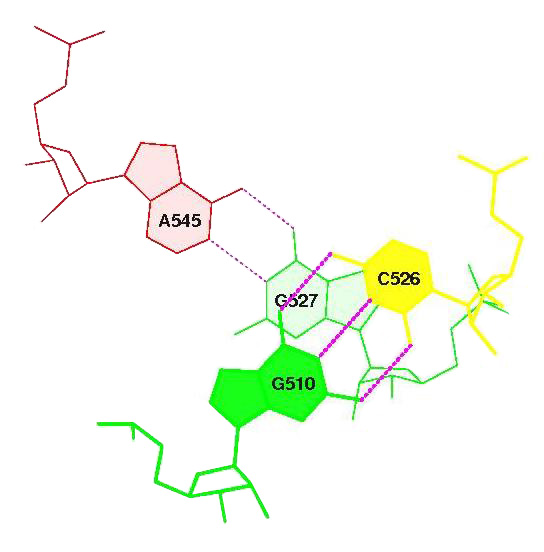
D-стебель - антикодоновый стебель (перекрывание 1.16) 526-510, 545-527
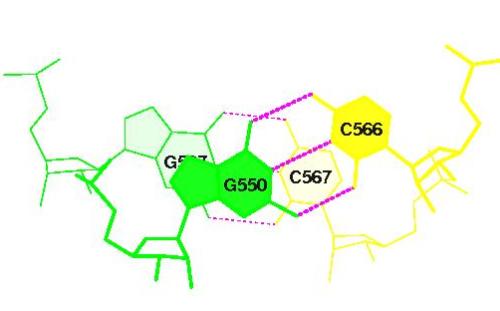
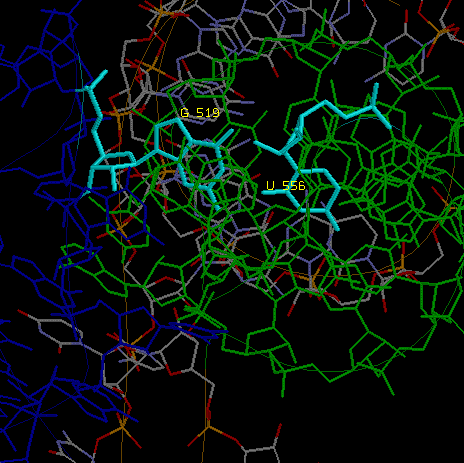
2. Дополнительные водородные связи между основаниями D- и Т-петель: G519-U556
Предсказание вторичной структуры тРНК
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted threshold=10 gap penalty=12 | Результаты предсказания по алгоритму Зукера (P не указан) |
| Акцепторный стебель | 5' 501-507 3' 5' 567-573 3' 7 пар |
все 7 | все 7 |
| D-стебель | 5' 510-513 3' 5' 523-526 3' 4 пары |
все 4 | все 4 |
| T-стебель | 5' 550-555 3' 5' 559-566 3' (560-561 - выпетливание) 6 пар |
ни одной | 5 из 6 |
| Антикодоновый стебель | 5' 527-533 3' 5' 539-545 3' 7 пар |
5 из 7 | 5 из 7 |
| Общее число канонических пар нуклеотидов | 21 | 15 | 19 |
Чтобы получить в einverted номера остатков стволов кроме акцепторного (самый большой вес), я вырезала из .fasta-файла участки последовательности 501-507 и 567-574. Это позволило программе обнаружить ещё два ствола. При попытках изменять параметры без вырезания вышеуказанных остатков программа либо показывала только один ствол из 7 пар нуклеотидов, либо (при очень низких штрафах за гэп) выравнивала половины последовательности друг с другом без разделения на стволы с большим количество гэпов (один ствол с многочисленными выпетливаниями), что не имело биологического смысла.
Наиболее близкое к реальности изображение было выдано программой mfold буквально при первом запуске. При увеличении P до 25 (количества результатов - до 24) ни одного более адекватного результата получено не было.

|