Ресеквенирование. Поиск полиморфизмов у человека.
Задание 1
Дано:
1. Чтения экзома, картирующиеся на участок хромосомы человека (chr3.fastq)
2. Хромосома человеческого генома (chr3.fasta)
Анализ качества чтений
С помощью программы FastQC был выполнен контроль качества чтений. FastQC принимает на фход файл с ридами, проволит анализ качества и приводит обширный отчет, состоящий из нескольких модулей. Для этого использовалась следующая команда:
fastqc chr3.fastq
В результате был получен архив (chr3_fastqc.zip), содержащий html-файл (chr3_fastqc.html), который, собственно, и представляет отчет.
В отчете содержится множество информации и графиков. График "Per base quality" приведен на рис.1.
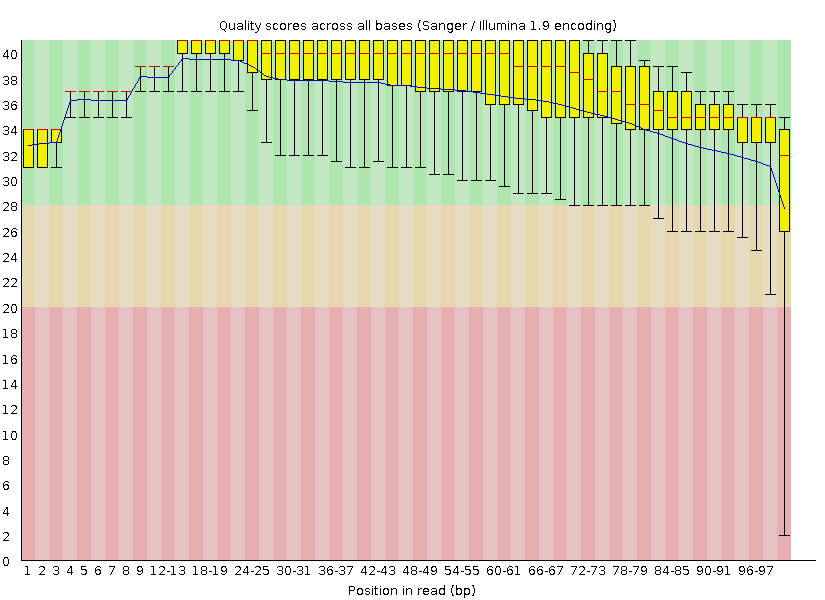
Рис. 1. Качетво чтений до очистки (FastQC)
По оси OX - позиция нуклеоитда в риде (от 1 до 100), по оси OY - параметр качества. Чем выше показатель качества, тем меньше вероятность ошибки. Например, для значения параметра качества 30 вероятность ошибки равна 0,001(точность 99,9%). Желтые прмоугольники на графике отражают интерквантильный размах(разница между верхней и нижней квартилями), отходящие от них "усы" отражают размах между 10-й и 90-й процентилями. Кроме того, синяя линия отражает медиану, красная - среднее значение. Все поле графика разделено на 3 части (зеленого, желтого и красного цветов), по которому можно легко оценить качество рида.
Итак, исходный рид имеет довольно хорошее качество, за исключением последних нуклеотидов, "усы" которых достигают красной зоны. В связи с этим была проведена очистка ридов от концевых нукдеотидов с низким качеством.
Очистка чтений
С помощью программы Trimmomatic с конца каждого чтения быи отрезаны нуклеотиды с качеством ниже 20. Причем после данной процедуры были отсавлены только чтения длиной не меньше 50 нуклеотидов.
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr3.fastq outfile.fastq TRAILING:20 MINLEN:50
Функция TRAILING отрезает с конца рида нуклеотиды с качеством ниже заданного (в данном случае - ниже 20), а MINLEN задает минисальную длину рида, который необходимо оставить.
На рис.2 приведен график качества чтений после очистки.
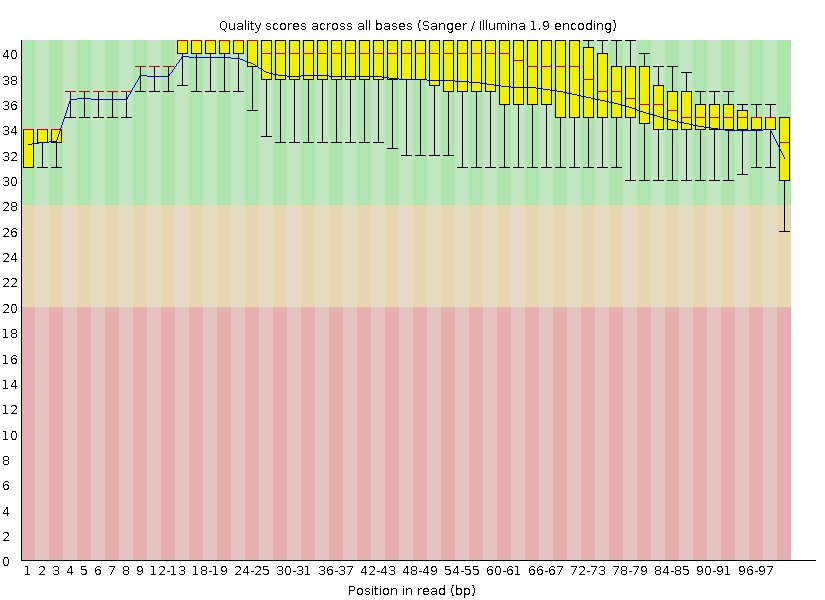
Рис. 2.Качетво чтений после очистки (FastQC)
До очистки было 20932 последовательности и их длины лежали в диапазоне 30-100. После очистки осталось 20570 последовательностей с длинами 50-100.
Еще один график, содержащийся в отчете FastQC представлен на рис. 3 и 4. Он отражает распределение среднего качества среди чтений.
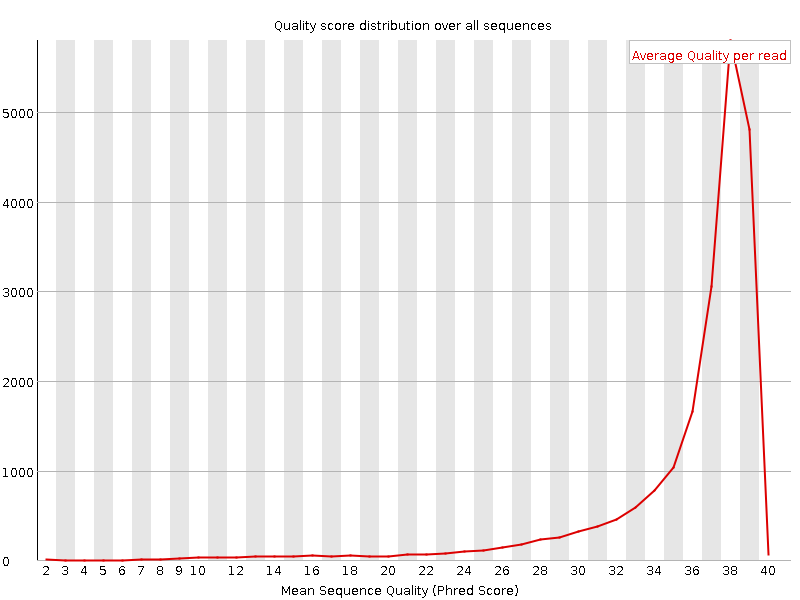
Рис. 3.Среднее качество среди чтений до очистки (FastQC)
Анализируя рис. 3 можно заключить, что до очистки большая часть ридов имеет качество >30. Однако имеются и риды, качество которых ниже 20. Их довольно мало, однако они все же есть. Но уже после очистки ридов с качеством ниже 20 уже нет, и это можно увидеть на рис. 4. Также можно заметить, что и после очистки большинство ридов имеет качество >30.
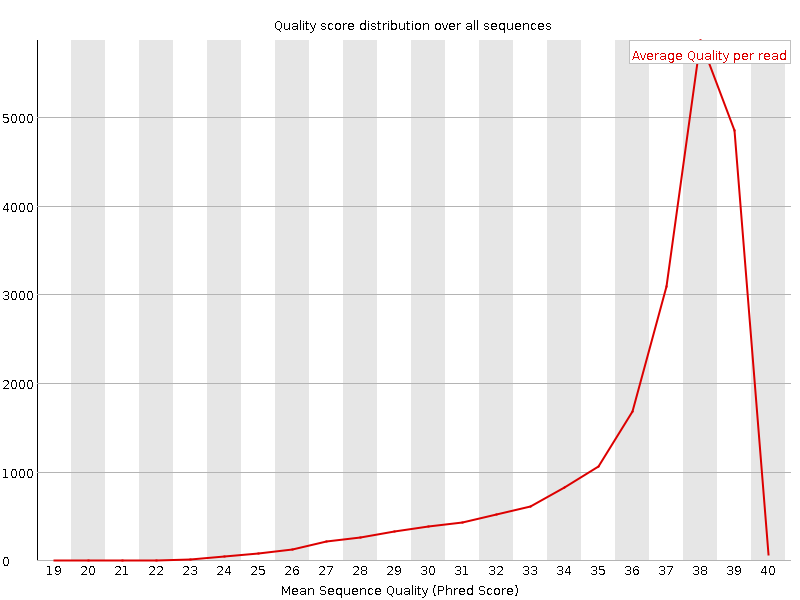
Рис. 4.Среднее качество среди чтений после очистки (FastQC)
Картирование чтений
С помощью программы BWA были откартированы очищенные чтения. BWA имеет 3 алгоритма: BWA-backtrack, BWA-SW и BWA-MEM. BWA-MEM - самый новый, рекомендован для картирования последовательностей с высоким качеством, благодаря чему работает более быстро и точно.
Сначала референсная последовательность была проиндексирована:
bwa index chr3.fasta
Далее было построено выравнивание прочтений и референса в формате .sam:
bwa mem chr3.fasta outfile.fastq > align.sam
Анализ выравнивания
Полученное выравнивание было проанализировано с помощью пакета samtools.
Сначала выравнивание чтений с референсом было переведено в бинарный формат .bam:
samtools view -b -o align.bam align.sam
Потом выравнивание чтений с референсом (получившийся после картирования .bam файл) было отсортировано по координате в референсе начала чтения:
samtools sort -T/tmp/align_sorted -o align_sorted.bam align.bam
После этого отсортированный .bam файл был проиндексирован и с помощью команды samtools idxstats было выяснено, сколько чтений откартировалось на геном:
samtools index align_sorted.bam samtools idxstats align_sorted.bam
Оказалось, что 20569 чтений откартировалось на хромосому, Число чтений, не картированных на хромосому: 1.
С помощью команжы depth была вычислено покрытие для каждого из нуклеотидов (depth.txt):
samtools depth align_sorted.bam > depth.txt
Далее из этого списка был выбран один из нуклеотидов (его координата - 52721621, покрытие - 280) и с помощью геномного браузера NCBI для данной хромосомы (сборка генома hg19 был найден экзон, в котором находится этот нуклеотид. Координаты экзона: 52721518-52721631.
После этого были получены покрытия нуклеотидов этого экзона(depth_exon.txt):
samtools depth -r chr3:52721518-52721631 align_sorted.bam > depth_exon.txt
Среднее покрытие нуклеотидов данного экзона составило 218,2105.
Поиск SNP и инделей
Сначала был создан файл с полиморфизмами в формате .bcf:
samtools mpileup -uf chr3.fasta align_sorted.bam > snp.bcf
Далее с помощью пакета bcftools был создан файл со списком отличий между референсом и чтениями в формате .vcf:
bcftools call -cv snp.bcf > snp.vcf
В нем всего содержалось 235 полиморфизмов, из которых 18 инделей и 217 замен.
| Координата | Тип полиморфизма | В референсе | В чтениях | Глубина покрытия | Качество чтений |
| 41939781 | Замена | G | A | 37 | 167.009 |
| 41996275 | Замена | A | G | 47 | 225.009 |
| 41923743 | Делеция | caaaa | caa | 5 | 129.467 |
У двух замен из табл.1 достаточно высокие показатели покрытия и качества, в отличие от делеции, покрытие которой составляет 5.
Аннотация SNP
Для анноттации SNP использовалась программа annovar. Поиск проводился по следующим базам данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
Программа annovar использует файлы в формате .avinput, поэтому сначала файл .vcf (только с snp, индели были удалены) был переведен в соответствующий формат:
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_only.vcf -outfile snp.avinput
После выполнения этой команды в файл snp.avinput было записано 217 SNP, из которых 121 транзиция и 96 трансверсий.
Далее полученные snp были проаннотированы с использованием различных баз данных:
perl /nfs/srv/databases/annovar/annotate_variation.pl -geneanno -dbtype refGene -buildver hg19 snp.avinput -outfile snp_refgene /nfs/srv/databases/annovar/humandb/ perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype snp138 -buildver hg19 snp.avinput -outfile snp_snp138 /nfs/srv/databases/annovar/humandb/ perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 snp.avinput -outfile snp_1000g2014oct /nfs/srv/databases/annovar/humandb/ perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -dbtype gwasCatalog -buildver hg19 snp.avinput -outfile snp_gwas /nfs/srv/databases/annovar/humandb/ perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 snp.avinput -outfile snp_clinvar /nfs/srv/databases/annovar/humandb/
1. RefGene
После применения соответствующей команды было получены следующие файлы: snp_refgene.log (содержит информацию о процессе выполнения аннотации), snp_refgene.variant_function (содержит описание SNP), snp_refgene.exonic_variant_function (содержит описание SNP, попавших в экзоны). В файле, содержащем описание всех SNP имеется разделение SNP на несколько групп: входящие в интроны, экзоны, UTR5 и UTR3 области. Количество SNP, попавших в каждую из групп приведено в табл. 2.
| Категория | Количество SNP |
| exonic | 13 |
| intronic | 198 |
| UTR3 | 4 |
| UTR5 | 2 |
Из всех SNP, 10 попало в ген ULK4, 3 - в ген FNDC3B.
2. dbSNP
В результате выполнения соответствующей команды было снова получено 3 файла: snp_snp138.log(содержит информацию о процессе выполнения аннотации), snp_snp138.hg19_snp138_dropped (содержит информацию об SNЗ, имеющих идентификатор в базе данных однонуклеотидных полиморфизмов - rs), snp_snp138.hg19_snp138_filtered (содержит SNP, не имеющих идентификаторов rs).
Таким образом было выяснено, что 178 SNP имеют rs идентиикатор, а 39 - не имеют.
3. 1000 genomes
Было получено 3 файла: snp_1000g_2014oct.log, snp_1000g_2014oct.hg19_ALL.sites.2014_10_dropped, snp_1000g_2014oct.hg19_ALL.sites.2014_10_filtered. Оказалось, что в этой базе аннотировано 175 SNP, а оставшиеся 42 SNP не аннотированы.
4. Gwas
Было получено 2 файла: snp_gwas.log, snp_gwas.hg19_gwasCatalog. Во втором файле содержалось 4 SNP, а так же признаки, с которыми они связаны: рост, артериальное давление, уровень адипонектина.
5. Clinvar
Было получено 3 файла: snp_clinvar.log, snp_clinvar.hg19_clinvar_20150629_dropped, snp_clinvar.hg19_clinvar_20150629_filtered. Оказалось, что все SNP попали в последний файл, то есть ни один из SNP не был аннотирован в этой базе данных.
Сводная таблица со аннотациями SNP по всем базам данных: snp.xlsx.
Ниже представлена сводная таблица со всеми командами, которые были использованы при выполнении данного задания.
| Команда | Пояснение |
| fastqc chr3.fastq | Анализ качества чтений |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr3.fastq outfile.fastq TRAILING:20 MINLEN:50 | Очистка чтений |
| bwa index chr3.fasta | Индексирование референсной последовательности |
| bwa mem chr3.fasta outfile.fastq > align.sam | Выравнивание очищенных чтений с проиндексированной референсной последовательностью |
| samtools view -b -o align.bam align.sam | Перевод выравнивания в бинарный формат |
| samtools sort -T/tmp/align_sorted -o align_sorted.bam align.bam | Сортировка выравнивания по координате начала чтения в референсной последовательности |
| samtools index align_sorted.bam | Индексирование отсортированного файла |
| samtools idxstats align_sorted.bam | Подсчет чтений, откартировавшихся на геном |
| samtools depth align_sorted.bam > depth.txt | Вычисление покрытия для каждого из нуклеотидов |
| samtools depth -r chr3:52721518-52721631 align_sorted.bam > depth_exon.txt | Получение покрытия для нуклеотидов одного из экзонов |
| samtools mpileup -uf chr3.fasta align_sorted.bam > snp.bcf | Создание файла с полиморфизмами |
| bcftools call -cv snp.bcf > snp.vcf | Создание файла с отличиями между референсом и чтениями |
| perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_only.vcf -outfile snp.avinput | Создание файла для annovar из .vcf файла |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -geneanno -dbtype refGene -buildver hg19 snp.avinput -outfile snp_refgene /nfs/srv/databases/annovar/humandb/ | Аннотация по базе данных RefGene |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype snp138 -buildver hg19 snp.avinput -outfile snp_snp138 /nfs/srv/databases/annovar/humandb/ | Аннотация по базе данных dbsnp |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 snp.avinput -outfile snp_1000g2014oct /nfs/srv/databases/annovar/humandb/ | Аннотация по базе данных 1000 genomes |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -dbtype gwasCatalog -buildver hg19 snp.avinput -outfile snp_gwas /nfs/srv/databases/annovar/humandb/ | Аннотация по базе данных GWAS |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 snp.avinput -outfile snp_clinvar /nfs/srv/databases/annovar/humandb/ | Аннотация по базе данных Clinvar |