Нуклеотидный blast
Задание 1
Требовалось определить таксономию и функцию последовательности, полученной в практикуме №6. Для этого был запущен blastn с данной последовательностью.
Параметры поиска: алгоритм blastn (Somewhat similar sequences), база данных Nucleotide collection (nr/nt), максимальное количество находок - 20000, остальные параметры - по умолчанию.
Результаты:
Было найдено 20000 находок. E-value худшей находки - 9e-75, лучшей - 0.0. Identities - 92%, лучшей - 98%.
Искомая последовательность является частичной последовательностью гена, кодирующего 18s rRNA (18s рибособная РНК). Эта РНК является структурным компонентом малых субъединиц рибосом эукариот. Нуклеотидная последовательность гена 18s rRNA является частым объектом секвенирования, так как ее испольуют для построения филогенетических деревьев.[1]
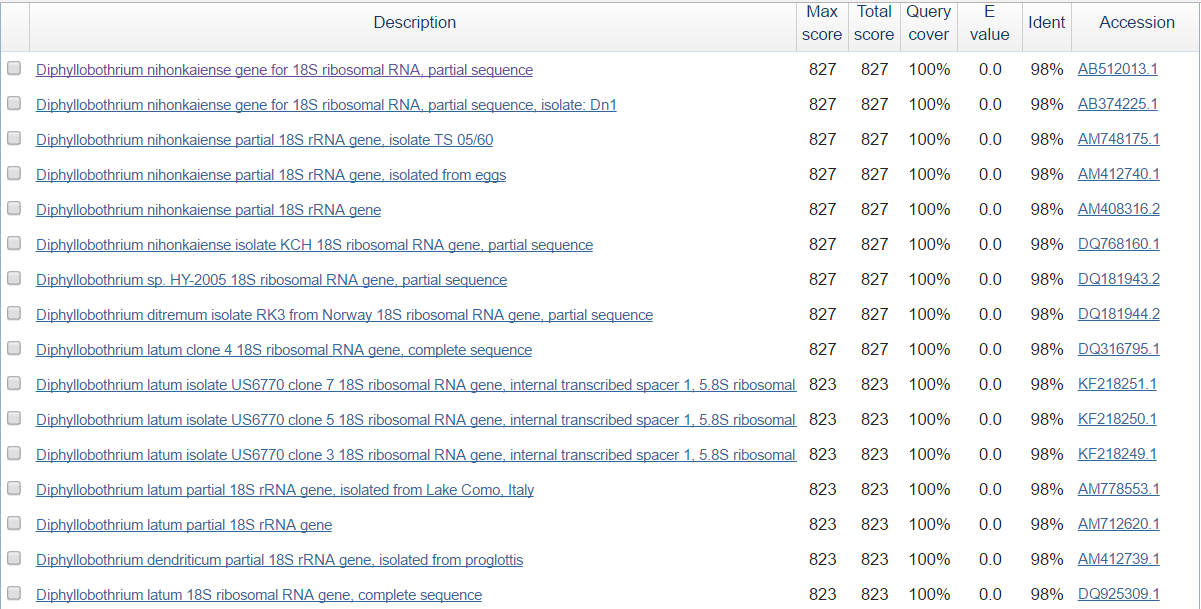 | |
| Рис. 1. Результаты применения blastn (первые несколько находок) |
Для определения таксономического положения была использвана функция BLAST Taxonomy Report (рис.2). У шести представителей рода Diphyllobothrium одинаковый Score: 827. Уровень сходства D.nihonkaiense, Diphyllobothrium sp., D.ditremum, D.latum, D.dendriticum с искомой последовательностью тоже одинаков: 2 замены на 100 п.н.
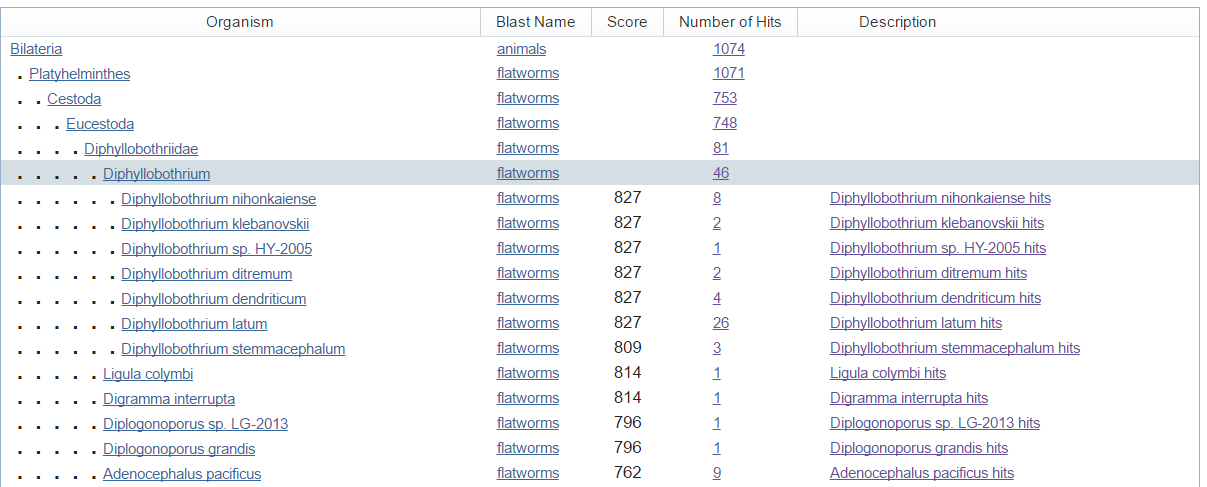 | |
| Рис. 2. Результаты BLAST Taxonomy Report |
Чтобы понять, на сколько нуклеотидов в среднем отличаются два разных вида друг от друга было запущено парное выравнивание участка 18s rRNA двух организмов: D.nihonkaiense (AB512013.1) и D.ditremum(DQ181944.2). Уровень сходства последовательностей - меньше 2 замен на 100 п.н.(identities - 99%). Аналогичное выравнивание было запущено для разных родов: Duthiersia(AF267290.1) и Diphyllobothrium(AB512013.1). Уровень сходства: 7 замен на 100 п.н.
Таким образом, нельзя точно определить, к какому виду принадлежит участок отсеквенированной последовательности. Однако до рода таксономическое положение определить возможно:
Раздел Bilateria Тип Platyhelminthes (Плоские черви) Класс Cestoda (Ленточные черви) Подкласс Eucestoda Семейство Diphyllobothriidae Род DiphyllobothriumНа рис.3 изображен представитель этого рода - Diphyllobothrium latum (Широкий лентец).
Задание 2
Требовалось сравнить списки находок нуклеотидной последовательности 3-я разными алгоритмами blast. Искомая последовательность была взята та же, что и в 1-м задании. Параметры поиска были немного изменены: база данных Nucleotide collection (nr/nt), максимальное количество находок - 500, таксон Diphyllobothriidae (taxid:28843), остальные параметры - по умолчанию.
| Алгоритм | Число находок | Query cover худшей находки, % | E-value худшей находки | Сходство худшей находки, % |
| blastn | 150 | 5 | 6.0 | 92 |
| discontiguous megablast | 129 | 11 | 5e-07 | 84 |
| megablast | 95 | 6 | 1e-04 | 97 |
Данные различия обусловлены тем, что в качестве "затравки" выравнивания эти алгоритмы берут разное число нуклеотидов:
На рис. 4 показаны худшие находки blastn, которые не попали в списки discontiguous megablast и megablast.
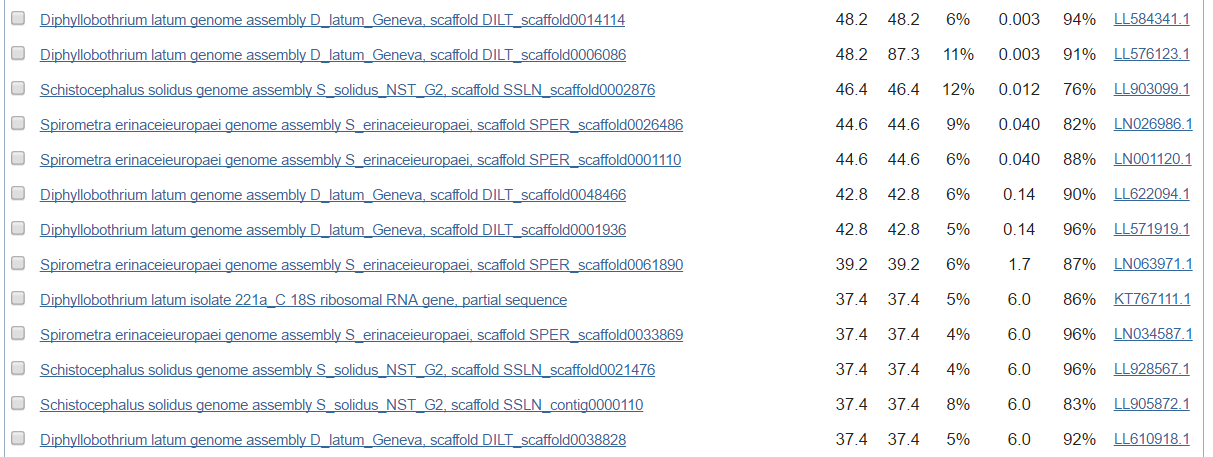 | |
| Рис. 4. Худшие находки blastn |
Таким образом, данные три алгоритма имеют разные области применения. Если задача состоит в том, чтобы найти наиболее близких гомологов - стоит применить megablast, discontiguous megablast позволяет находить гомологов,имкющих довольно схожие фрагменты, между которыми встроены немного различающиеся участки. Больше всего последовательностей находит blastn, однако далеко не все их них являются гомологами исходной последовательности.
Задание 3
Требовалось проверить наличие гомологов трех белков в геноме Amoeboaphelidium protoccocarum. Выбранные белки представлены в табл.2.
| Имя записи в UniProt | Название белка | Описание |
| HSP7C_HUMAN | Heat shock cognate 71 kDa protein | консервативный шаперон HSP70, белок теплового шока; имеется у большинства организмов из всех царств |
| TERT_HUMAN | Telomerase reverse transcriptase | теломераза,восстанавливающая длину хромосомы при репликации; имеется у большинства (но не всех) эукариот |
| RPB1_HUMAN | DNA-directed RNA polymerase II subunit RPB1 | субъединица ДНК-зависимой РНК-полимеразы II |
Для выполнения этого задания была использована локальная версия BLAST+, алгоритм - tblastn. Сначала была создана локальная база данных по сборке генома X5 (Amoboeaphelidium protoccocarum) с помощью команды makeblastdb. Аминокислотные последовательнотсти белков были собраны в одном файле. Далее был запущен tblastn. Ссылка на выходной файл: x5.out.
| Белок | Число находок | Запись с лучшей находкой | Query cover лучшей находки | E-value лучшей находки | Идентичность лучшей находки |
| HSP7C_HUMAN | 22 | scaffold-199 | 93,9% | 0.0 | 78.09% |
| TERT_HUMAN | 3 | scaffold-17 | 50,26% | 8e-23 | 26.58% |
| RPB1_HUMAN | 21 | scaffold-300 | 78,5% | 0.0 | 51,65% |
Исходя из полученных данных можно сделать следующте выводы:
1. Гомолог белка HSP7C_HUMAN есть у Amoeboaphelidium protoccocarum. Это неудивительно, так как последовательность этого белка является довольно консервативной и он встречается во всех царствах.
2. Гомолог белка TERT_HUMAN предположительно тоже есть у Amoeboaphelidium protoccocarum, однако параметры его сходства гораздо ниже. Это может быть обусловлено тем, что лишь часть доменов являются консервативными. Возможно также, что белок частично утратил свою функцию.
3. Гомолог белка RPB1_HUMAN есть у Amoeboaphelidium protoccocarum.
Задание 4
Требовалось найти один ген белка, закодированный в одном контиге Amoboaphelidium.
Сначала был выбран подходящий контиг длины порядка десятков тысяч пар нуклеотидов с помощью команды infoseq пакета EMBOSS. Далее была вырезан scaffold-499 длиной 32750 п.н. с помощью команды seqret. Полученный fasta-файл был использован в качестве запроса в blastx (база данных - RefSeq). Первые несколько находок представлены на рис.5.
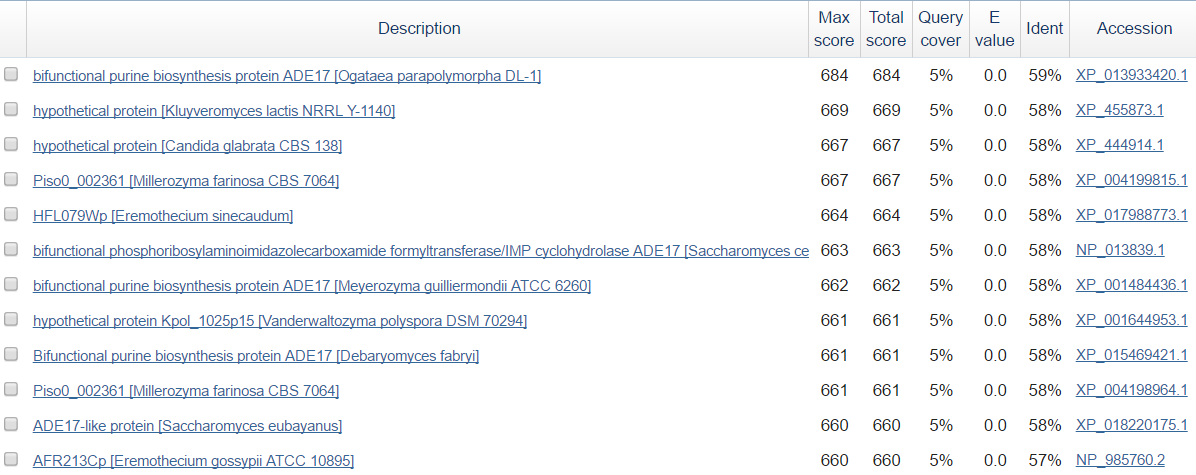 | |
| Рис. 5.Результы примениения blastx к scaffold-499 |
Исходя из полученных результатов можно сделать вывод о том, что в данном контиге расположен ген, кодирующий белок, который связан с биосинтезом пуринов (bifunctional purine biosynthesis protein ADE17). Наиболее изучен этот белок у Saccharomyces cerevisiae - гены ADE16 и ADE17 кодируют 5-аминоимидазол-4-карбоксамид -рибонуклеотид изоферменты трансформилазы, катализирующие предпоследний шаг биосинтеза пуриновых нуклеотидов.[2]
Задание 5
В этом задании нужно было классифицировать геномы родственных вирусов по сходству последовательностей. Для этого были выбраны 2 штамма свиного бокавируса, 2 штамма бокавируса человека и 1 штамм бокавируса крысы (все эти вирусы относятся к одному роду Bocaparvovirus):
Геномы всех организмов были загружены и сохранены в одном fasta-файле. С помощью команды makeblastdb была создана база для этого fasta файла. Далее был применен tblastx: поиск трансляций входных последовательностей против трансляций нуклеотидной базы данных. К полученному файлу был применен python-скрипт, который удаляет неинформативные и слабо сходные находки из полученной таблицы (параметры: identity - 65, max e-value 0.001). В итоге была получена excel-таблица.
Сходство между вирусами оценивалось по следующим критериям:
Результаты расчетов приведены в табл.4.
| Вирусы | по максимальному проценту сходства двух трансляций | по среднему проценту сходства всех трансляций | по суммарной длине сходных белковых последовательностей |
| GQ455987.1 и GQ455988.1 | 100% | 99,24% | 34329 |
| GQ455987.1 и KX017193.1 | 68.18% | 66,03% | 1695 |
| GQ455987.1 и NC_016647.1 | 68.57% | 68,57% | 104 |
| GQ455987.1 и NC_029133.1 | 76.19% | 69,76% | 509 |
| GQ455988.1 и KX017193.1 | 68.18% | 66,03% | 1695 |
| GQ455988.1 и NC_016647.1 | 68.57% | 68,57% | 104 |
| GQ455988.1 и NC_029133.1 | 76.19% | 56,25% | 509 |
| KX017193.1 и NC_029133.1 | 66.67% | 65,98% | 322 |
По данным результатам иожно сказать следующее:
Ссылки