EMBOSS: пакет программ для анализа последовательностей
Задание 1
1. Несколько файлов в формате fasta собрать в единый файл
Исходные файлы: ecoli1.fasta, ecoli2.fasta
ls *.fasta >list seqret @list seq1.fasta
Результат: seq1.fasta
2. Перевести выравнивание и из fasta формате в формат .msf
Исходные файлы: alignment.fasta
seqret alignment.fasta msf::alignment.msf
Результат: alignment.msf
3. Из файла с хромосомой в формате .gb вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле
Сначала был создан файл list со следующим содержимым:
genbank:AB032246[1:10] genbank:AB032246[30:76] genbank:AB032246[800:830]
Далее применена следующая команда:
seqret @list seq2.fasta
Результат: seq2.fasta
4. Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы
Исходный файл: seq1.fasta
seqretsplit seq1.fasta seq1out.fasta
Результат: ab214865.1.fasta, e09502.1.fasta
5. Транслировать данную нуклеотидную последовательность в шести рамках.
Исходный файл: seq1.fasta
transeq ecoli1.fasta -frame=6 transl.fasta
Результат: transl.fasta
6. Транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода. Результат - в одном fasta файле.
Исходный файл: seq1.fasta
transeq seq1.fasta -table 0 transl2.fasta
Результат: transl2.fasta
7. Выдать в выходной поток число совпадающих букв между второй последовательностью выравнивания и всеми остальными (на выходе только имя последовательности и число).
Исходный файл: alignment.msf
infoalign alignment.msf -refseq 2 -only -name -simcount alignment.infoalign
Результат: alignment.infoalign
8. Перевести аннотации особенностей в записи формата .gb в табличный формат .gff.
Исходный файл: seq3.gb
featcopy seq3.gb seq3.gff
Результат: seq3.gff
9.Из данного файла с хромосомой в формате .gb получить fasta файл с кодирующими последовательностями; (*) добавить в описание каждой последовательности функцию белка (из поля product)
Исходный файл: seq3.gb
extractfeat seq3.gb -type CDS -describe product
Результат: seq3_CDS.fasta
10.Перемешать буквы в данной нуклеотидной последовательности;
Исходный файл: ecoli1.fasta
shuffleseq ecoli1.fasta ecoli_shuffle.fasta
Результат: ecoli_shuffle.fasta
11.Найдите частоты кодонов в данных кодирующих последовательностях
Исходный файл: ecoli1.fasta
cusp ecoli1.fasta ecoli.cusp
Результат: ecoli.cusp
12.Найдите частоты динуклеотидов в данной нуклеотидной последовательности и сравните их с ожидаемыми
Исходный файл: ecoli1.fasta
compseq ecoli1.fasta -word 2 ecoli.compseq
Результат: ecoli.compseq
13. Выровняйте кодирующие последовательности соответственно выравниванию белков - их продуктов
Исходный файл: seq4.seq (нуклеотидные последовательности), seq4.fasta(белковые послежовательности)
tranalign seq4.seq seq4.fasta tranalign.fasta
Результат: tranalign.fasta
Задание 2
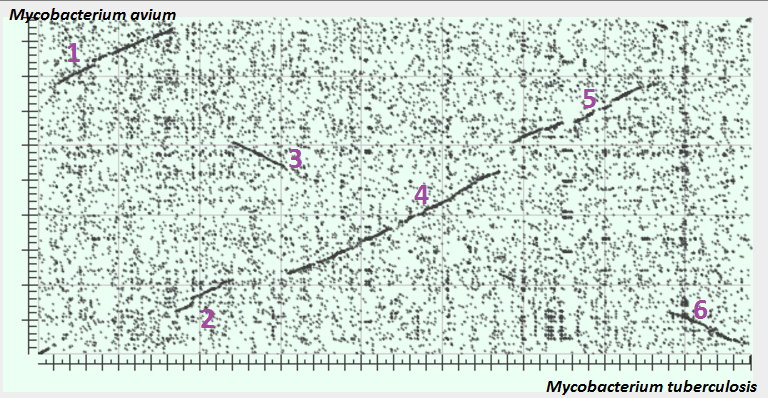
2A. Карта локального сходства Mycobacterium tuberculosis и Mycobacterium avium
Для сравнения были выбраны геномы бактерий и рода Mycobacterium: M.tuberculosis(NC_000962.3) и M. avium(NC_002944.2).
Был использован blast2seq, алгоритм blastn, на сайте NCBI. В результате было получено парное выравнивание геномов двух бактерий, а также карта локального сходства, представленная на рис.1.
 | |
| Рис.1.Карта локального сходства Mycobacterium tuberculosis и Mycobacterium avium |
На карте локального сзодства можно выделить несколько крупных эволюционных событий, произошедших с рассматриваемыми геномами бактерий:
Чтобы точно определить, что произошло в обласи №2 (вставка или делеция), нуклеотидная последовательность данной "вставки" была скопирована и запущен blastn по таксону Mycobacterium.
Возможные исходы:
Предполагалось, что должен быть найден очень похожий участок, поэтому сначала был запущен MEGABLAST. Все находки оказались из вида M. avium. Однако для того, чтобы убедиться в том, что очень похожих последовательностей нет, был запущен blastn (word size=15) Достоверных находок найдено мало. Они представлены на рис.2.
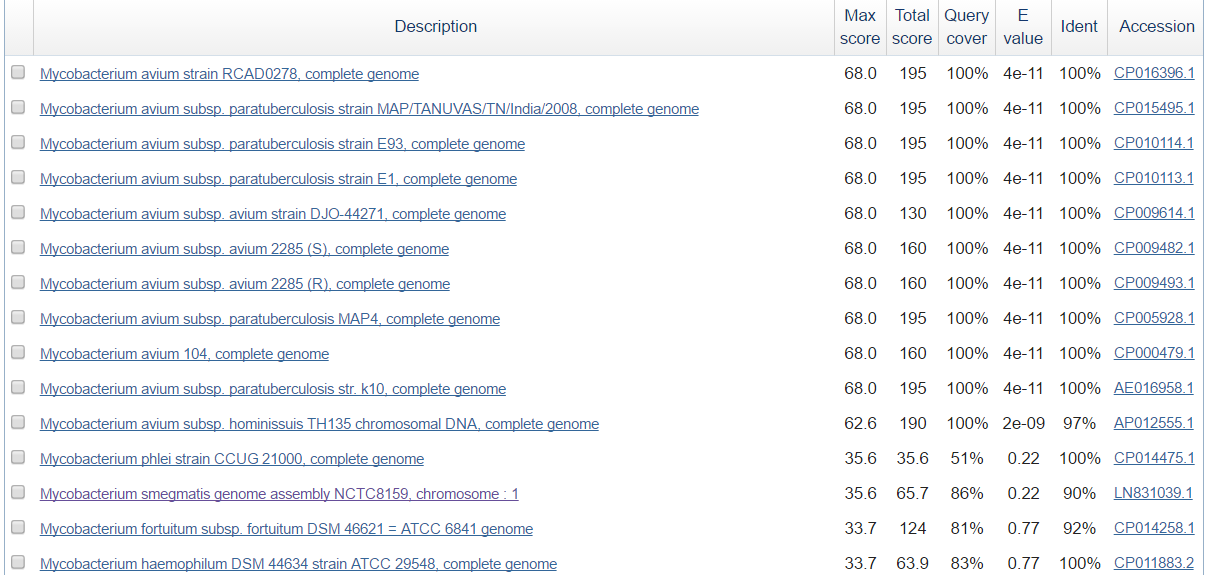 | |
| Рис.2.Результаты blastn по таксону Mycobacterium |
Вывод: скорее всего в области № 2 наблюдается вставка M. avium за счет горизонтального переноса в этот геном.
2B. Нуклеотидный пангеном штаммов бактерий вида Mycobacterium tuberculosis
Для выполнения этого задания были выбраны следующие штаммы:
Сначала была создана директория npg (elenavas/term3/block2/npg). В ней был создан файл genomes.tsv с информацией о вышеперечисленных последовательностях в специальном формате. Далее были выполнены следующие команды:
npge -g npge.conf npge Prepare npge Examine
Далее были изменены некоторые параметры в файле npge.conf, а именно: MIN_IDENTITY = Decimal('0.85') и WORKERS = 1 (рекомендованное значение MIN_IDENTITY: 0.899).
Протокол выполнения пангенома был записан в айл log:
npge MakePangenome > log
После этого была записана аналитическая информация о пангеноме:
npge PostProcessing
Описание синтеничных участков
Синтеничные области (g-блоки) - участки геномов, состоящие из ортологичных областей с сохранением их порядка на хромосоме для сравниваемых геномов. Они состоят из состоят из последовательно идущих во всех геномах s-блоков, перемежающихся блоками других типов (r-, h-, u- и m-). Список глобальных блоков содержится в global-blocks/blocks.gbi, полученном в результате PostProcessing. Для выполнения этого задания данные были скопированы в Excel, и удалены строки, не содержащие g-блоков.
На рис. 3 приведено выравнивание g-блоков. 3 блока (g4x2206, g4x2781459, g4x101) являются консервативными. Последовательности g-блоков у штаммов 49-02 и F11 одинаковые, как и у штаммов EAI5 и H37Rv. Эти пары штаммов отличаются порядком следования g-блоков, что говорит о том, что произошли транслокации.
 | |
| Рис.3.Выравнивание g-блоков |
Описание ядра пангенома
Ядро пангенома состоит из s-блоков, то есть из стабильных (коровых) блоков, которые присутствуют по одному в кадом геноме.
По объединенному выравниванию s-блоков было построено филогенетическое дерево, визуализация которого была проведена с помощью сервиса ITOL. Результыт представлен на рис.4. Примечательно, что несмотря на расположение g-блоков (по котоорому близкими оказались 49-02 и F11Б а также EAI5 и H37Rv), по объединенному выравниванию s-блоков другие пары штаммы (F11Б и H37Rv, а также 49-02 и EAI5 ) оказались близкими в эволюционном плане. Причем расхождение между 49-02 и EAI5 произошло несколько позже, чем между F11Б и H37Rv.
 | |
| Рис.4.Филогенетическое дерево, построенное по объединенному выравниванию s-блоков |
Описание повторов
Блок r20x526 состоит из 526 нуклеотидов. Во всех штаммах имеется по 5 копий этого блока. Процент консервативности составляет 99,61%. В визуализаторе qnpge отображается, что в данном блоке закодировано 20 генов(то есть по 5 на каждый штамм, или, что то же самое, 1 ген в 1 участке, который потом копируется 5 раз в каждом из штаммов).
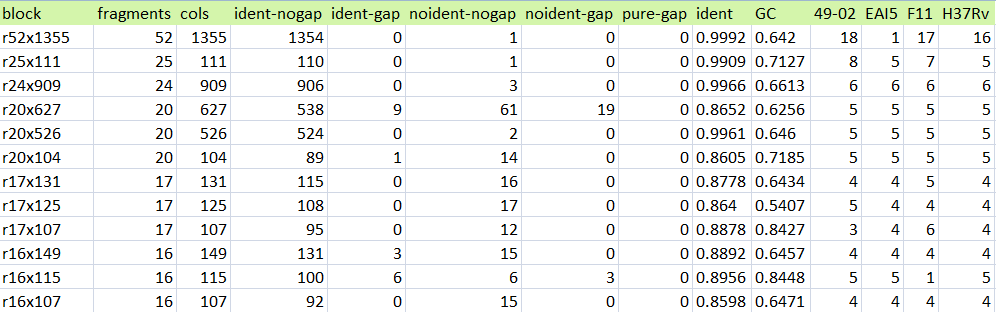 | |
| Рис.5.R-блоки(часть информации из файла pangenom.bi) |
Описание крупной делеции
Из файла pangenom.bi, часть которого представлена на рис. 6 был выбран h-блок (т.е. блок, который присутствует в части из анализируемых геномов) h3x1027. Как видно их рис. 6, данный блок есть у всех штаммов, кроме штамма 49-02. В данном блоке у этих штаммов закодированы следующие белки(информация из qnpge):
CDS M943_01475_M943_01475 hypothetical protein (EAI5), 2514 bp CDS TBFG_10287_TBFG_10287 PPE family protein (F11), 2514 bp CDS Rv0280_Rv0280 PPE family protein PPE3 (H37Rv), 2514 bp
Для проверки того, что у 49-02 действительно произошла делеция был запущен blastn с нуклеотидной послеловательностью белка и геномом штамма 49-02. Ни одной находки обнаружено не было. Следовательно, можно утверждать, что у 49-02 произошла делеция участка, кодирующего ген белка семейства PPE.
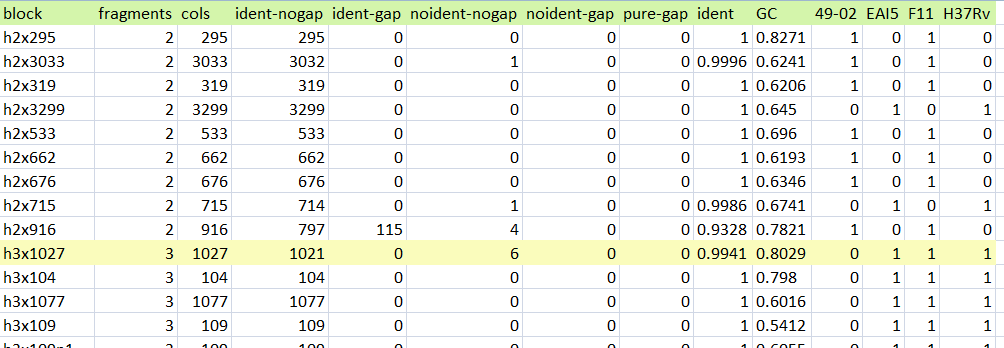 | |
| Рис.6. R-блоки (часть информации из файла pangenom.bi) |
Описание последовательности, имеющейся только в одном геноме
Уникальные последовательности из одного генома,у которой нет гомологов среди других геномов называются u-блоками. Примером такого u-блока может быть блок u1x6611, который имеется только у штамма F11. В данном участке содержатся 4 гена (первый не полностью), которые кодируют следующие белки(информация из qnpge):
CDS TBFG_11776_TBFG_11776 glycosyl transferase (F11), 1545 bp < (1985745-1986222) CDS TBFG_11777_TBFG_11777 hypothetical protein (F11), 1146 bp < (1986351-1987496) CDS TBFG_11778_TBFG_11778 membrane protein, mmpL family (F11), 1104 bp > (1987722-1988825) CDS TBFG_11779_TBFG_11779 transposase (F11), 2838 bp > (1989026-1991863)
Однако в базе данных GenBank указаны другие белки на данных позициях. Они представлены на рис.7
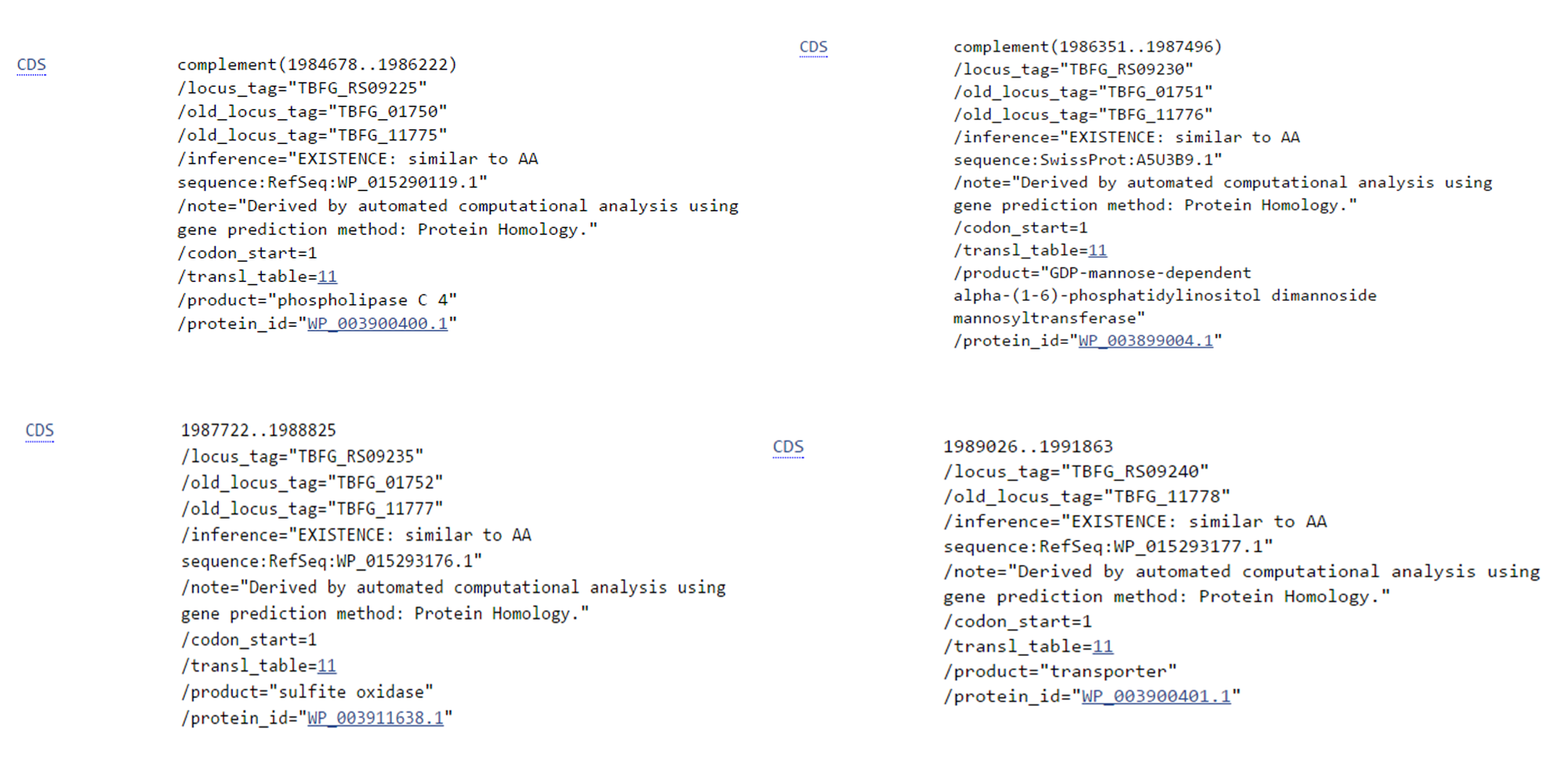 | |
| Рис.7. CDS уникального блока u1x6611 |